

Preparation and refining method of parecoxib and intermediate thereof
A technology for parecoxib and a synthesis method, applied in the field of medicinal chemistry, can solve problems such as substandard material purity, and achieve the effects of shortening working hours and saving consumption
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

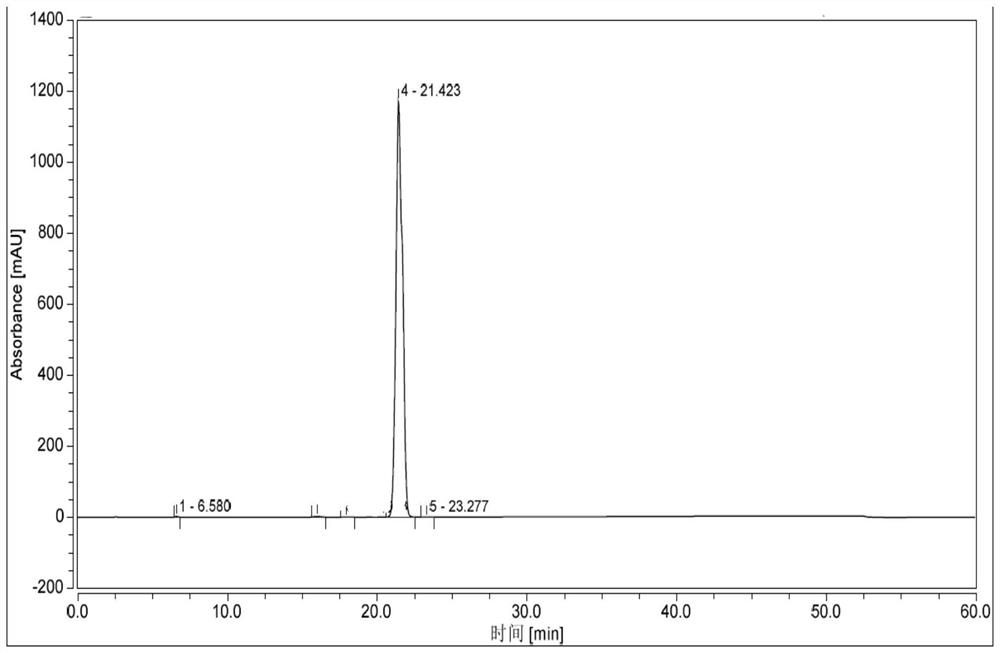
Examples
Embodiment 1
[0074] Embodiment 1 sulfonation step
[0075] Add 15.6kg of dichloromethane and 6.0kg of SM1 into the reactor, stir and cool down to 0-15°C. Slowly add 26.64kg of chlorosulfonic acid dropwise, and control the internal temperature to 0-15°C. After dropping, rise to 40-45°C to reflux for 4 hours. Lower the temperature to 0-15°C, slowly add the reaction solution dropwise to 72kg of purified water, and control the internal temperature to 0-30°C. Stand and separate the layers, separate the organic phase, extract the aqueous phase with 18 kg of ethyl acetate (no emulsification, no need to stand), separate the organic phase, and extract and wash with 5.0 kg of purified water. After concentrating the organic phase of dichloromethane, adding ethyl acetate, the organic phase was concentrated again until the remaining mass of the system was 15kg±1kg. Control the temperature at 30-40°C and add 33kg of n-hexane dropwise, slowly lower the temperature to 10-20°C, stir and crystallize for ...
Embodiment 2
[0076] Embodiment 2 sulfonation step
[0077] Add 15.6kg of dichloromethane and 6.0kg of SM1 into the reactor, stir and cool down to 0-15°C. Slowly add 26.64kg of chlorosulfonic acid dropwise, and control the internal temperature to 0-15°C. After dropping, rise to 40-45°C to reflux for 4 hours. Lower the temperature to 0-15°C, slowly add the reaction solution dropwise to 72kg of purified water, and control the internal temperature to 0-30°C. Stand to separate the layers, separate the organic phase, extract with 10kg*2 dichloromethane, and let stand for no less than 2 hours (repeat the extraction operation once). The organic phases were combined, washed twice with 2.0kg*2 purified water, dried over anhydrous sodium sulfate, and concentrated. Add 7.2kg of ethyl acetate, heat up to 30-30°C and stir to dissolve, slowly add 32.4kg of n-hexane dropwise, slowly cool down to 10-20°C, stir and crystallize for 1 hour, centrifuge, rinse with 6kg of n-hexane, 60-65 °C and dried under ...
Embodiment 3
[0082] Embodiment 3 amination step
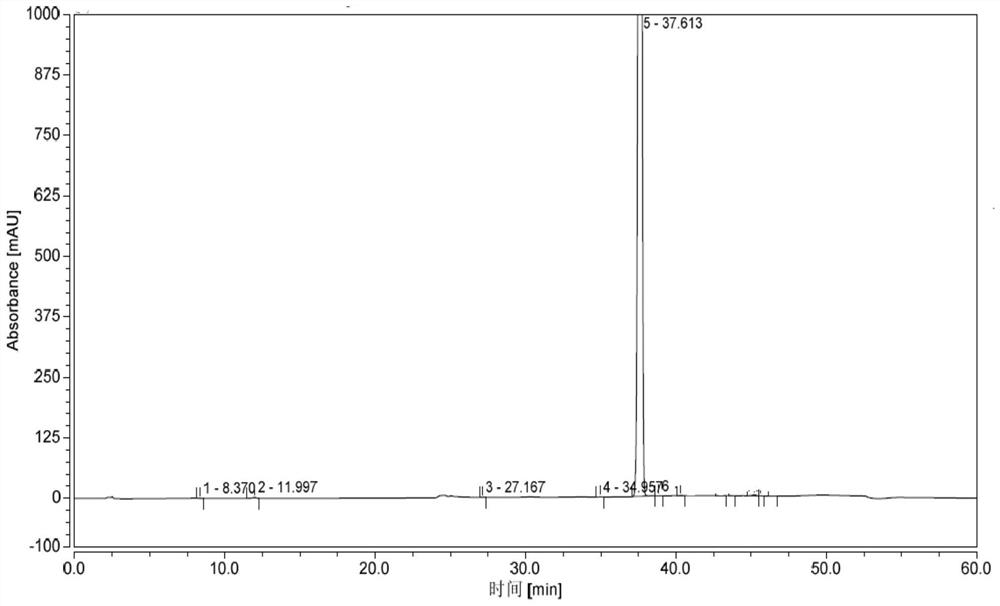
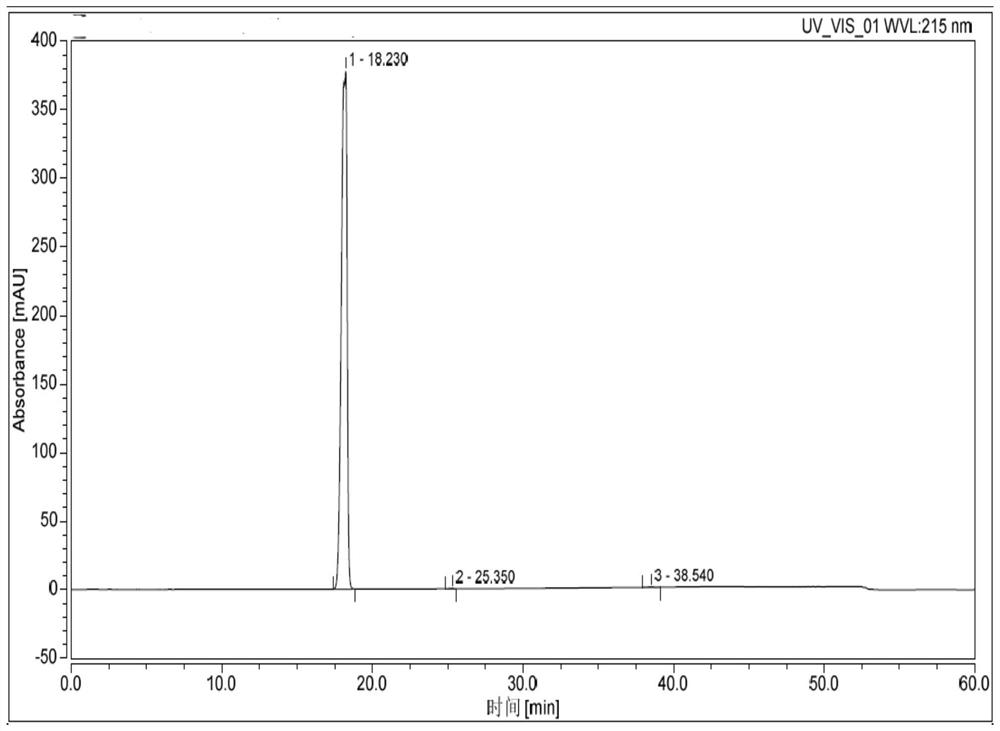
[0083] Add 20kg of ammonia water into the reactor, and add 6.0kg of intermediate I (prepared in Example 2) in dichloromethane solution (dissolve 6.0kg of intermediate I in 9.1kg of dichloromethane) dropwise under temperature control at 0-15°C. After dropping, the temperature was raised to 25-30°C to react for 1 hour. Add 28kg of purified water, and continue stirring at 25-30°C for 1 hour. Centrifuge, wash with purified water, and dry to obtain 6 kg of crude product. Add 30kg of ethanol with a water content of 5%, heat up to 70-80°C to dissolve, cool down to 20-25°C for crystallization for 1 hour, centrifuge, rinse with 1.2kg of absolute ethanol, and dry to obtain 5.73kg of white solid, namely for valdecoxib. Purity 99.6% (see attached figure 2 ).
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More