

Method for analyzing nucleic acid reactions
a nucleic acid and reaction technology, applied in the field of nucleic acid reaction analysis methods, can solve the problems of labor-intensive and expensive methods, and labor-intensive and labor-intensive methods for eukaryotic chromosomes
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

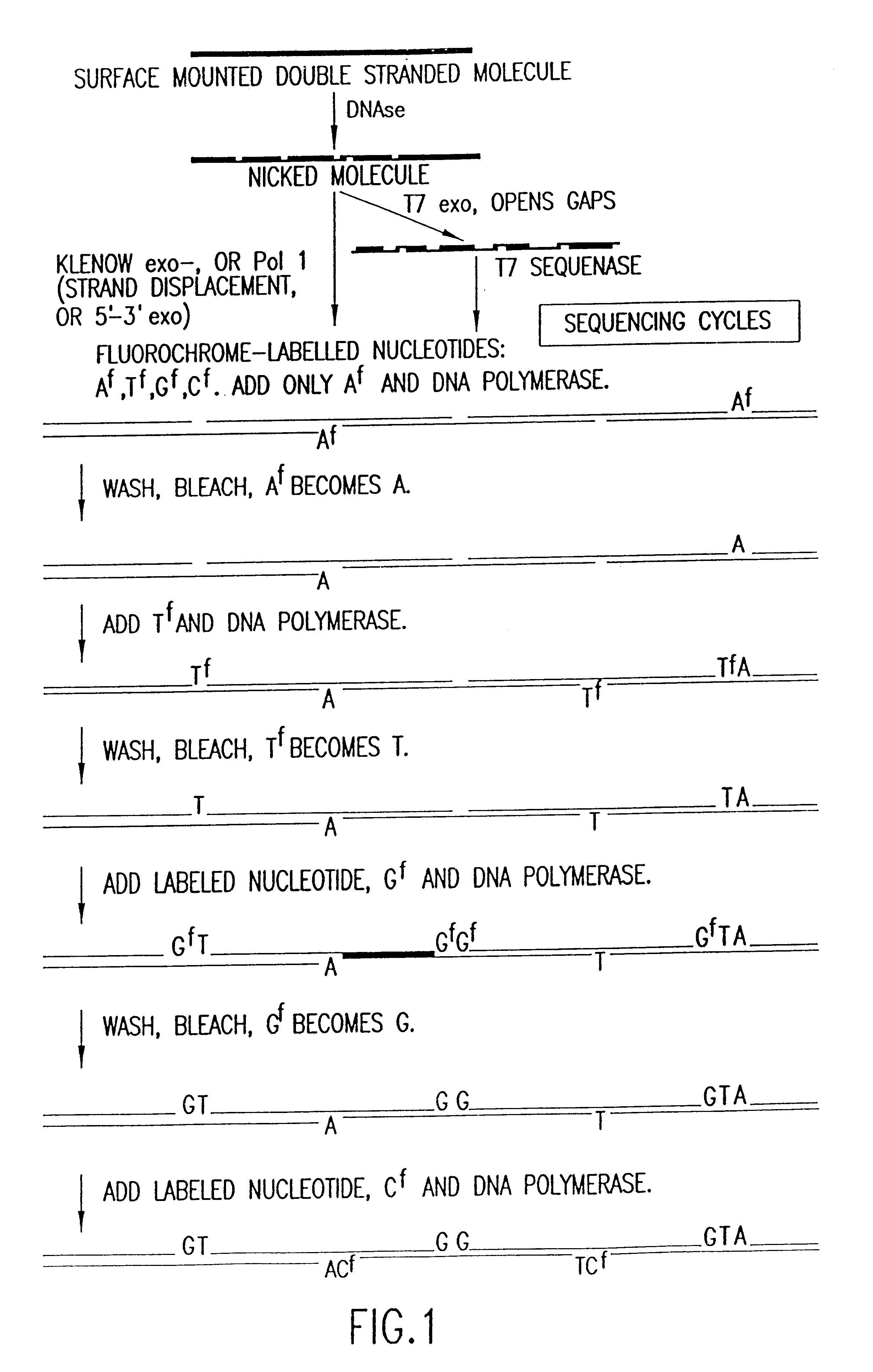
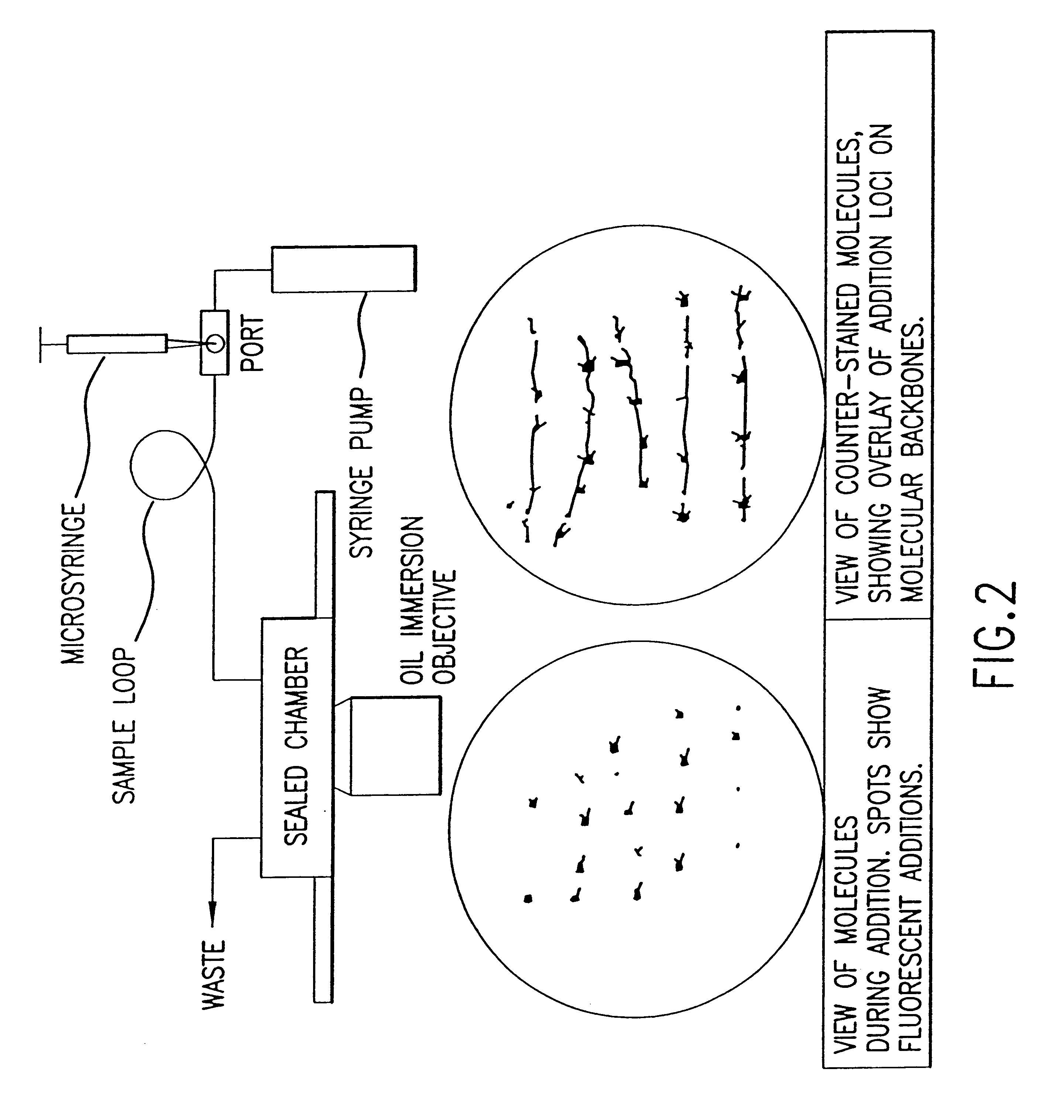

Examples
case 1
..fwdarw.p.sub.b
Taking the first partial derivative of the likelihood function with respect to p.sub.b gives: ##EQU12##
where p.sub.b is the probability that the data is invalid, and e.sub.j, d.sub.j are as defined in Eqn. (3). Taking the second partial derivative gives: ##EQU13##
In accordance with a preferred embodiment of the present invention .LAMBDA. can be optimized iteratively to estimate the best value of p.sub.b, by means of the following application of the Newton's equation: ##EQU14##
where the first and second partial derivatives are as indicated above. The above expression is used in the iterative optimization in accordance with a preferred embodiment of the present invention. Iterative techniques for function optimization are known in the art and need not be considered in detail.
case 2
..fwdarw..lambda..sub.n
The expected number of cuts per "bad" molecule is simply estimated to be the average number of cuts. Note that, ##EQU15##
should be zero at the local maxima. Thus a good approximation is obtained by taking ##EQU16##
leading to the update rule ##EQU17##
Thus, in accordance with a preferred embodiment of the present invention, .lambda..sub.n is simply the average number of cuts per molecule.
case 3
..fwdarw.h.sub.i, p.sub.ci, .sigma..sub.i (i=1, . . . N), or .lambda.
Unlike in the previous two cases, these parameters are in the innermost section of the probability density expression and computing any of these gradients will turn out to be computationally comparable to evaluating the entire probability density. In this case, ##EQU18##
as the relative probability density of the alignment A.sub.jk for data item D.sub.j.
Thus, the expression for the partial derivative with respect to .theta. simplifies to ##EQU19##
Before examining the updating formula for each parameter optimization, the following notations are introduced for future use. In a preferred embodiment, the quantities defined below are efficiently accumulated for a fixed value of the set of parameters.
.psi..sub.0i.ident..SIGMA..sub.j.SIGMA..sub.k.pi..sub.jk m.sub.ijk.ident. Expected number of cuts matching h.sub.i
.psi..sub.1i.ident..SIGMA..sub.j.SIGMA..sub.k.pi..sub.jk m.sub.ijk s.sub.ijk.ident. Sum of cut locations matchi...
PUM
| Property | Measurement | Unit |
|---|---|---|
| molecular length | aaaaa | aaaaa |
| contour length | aaaaa | aaaaa |
| temperature | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More