

Method for synthesizing 13C-labeled alpha-aminobutyric acid
A technology of aminobutyric acid and synthesis method, which is applied in chemical instruments and methods, carboxylate preparation, carboxylic acid amide preparation, etc., can solve the problems of inability to selectively synthesize carboxyl groups, unfavorable industrial production, and increased raw material cost, etc., and achieves good results. The effect of economy and practical application value, less by-products, and simple operation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
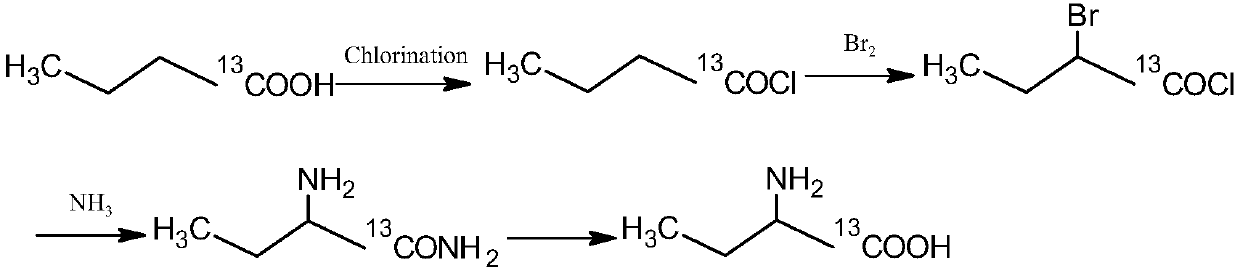
[0031] 13 The synthetic method of C-labeled α-aminobutyric acid adopts the following method:
[0032] 5g n-butyric acid-1-( 13 COOH) was dissolved in 50ml of 1,2-dichloroethane, 6.68g of thionyl chloride was added at 0°C, and the reaction was stirred at 0°C for 6h until no HCl gas was released. The reaction liquid spins off the solvent to obtain the intermediate intermediate (1) n-butyryl chloride- 13 C 5.70g.
[0033] 5.70g intermediate (1) n-butyryl chloride- 13 Dissolve C in dichloromethane, heat to 80°C, slowly add 42.50 g of dry liquid bromine dropwise under stirring, and continue stirring for 12 hours. After the reaction, the reactant was cooled to room temperature. Spin to remove the solvent, distill under reduced pressure, collect 150 ° C light yellow oily fraction intermediate (2) 2-bromobutyric acid chloride- 13 C 8.90g.
[0034] Dissolve 33.42g of urotropine in 100ml of ethanol, stir and mix. Dry ammonia gas was introduced at room temperature until the pH of...
Embodiment 2
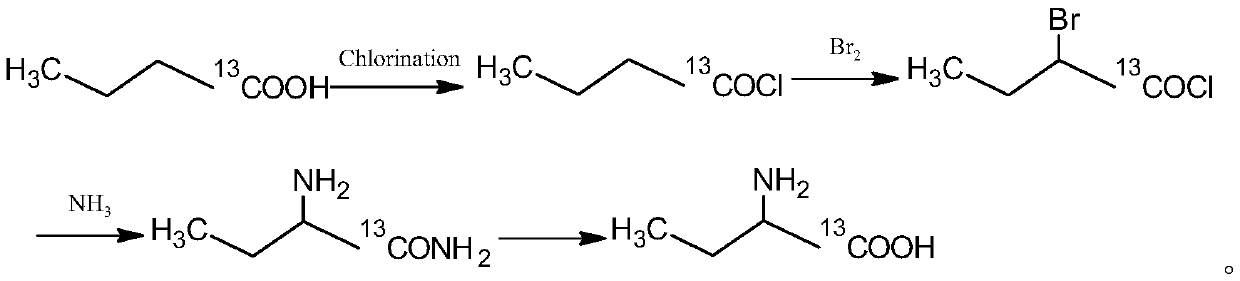
[0037] 13 The synthetic method of C-labeled α-aminobutyric acid adopts the following method:
[0038] 5g n-butyric acid-1-( 13 COOH) was dissolved in 50ml of dichloromethane, 20.00g of thionyl chloride was added at -20°C, and the reaction was stirred at 60°C for 8h until no HCl gas was released. The reaction liquid spins off the solvent to obtain the intermediate intermediate (1) n-butyryl chloride- 13 C 5.72g.
[0039] 5.72g intermediate (1) n-butyryl chloride- 13 Dissolve C in 1,2-dichloroethane, heat to 30°C, slowly add 8.50 g of dry liquid bromine dropwise under stirring, and continue stirring for 8 hours. After the reaction, the reactant was cooled to room temperature. Spin to remove the solvent, distill under reduced pressure, collect 150 ° C light yellow oily fraction intermediate (2) 2-bromobutyric acid chloride- 13 C 8.92g.
[0040] Dissolve 6.68g of urotropine in 100ml of methanol, stir and mix. Dry ammonia gas was introduced at room temperature until the pH ...
Embodiment 3
[0043] 13 The synthetic method of C-labeled α-aminobutyric acid adopts the following method:
[0044] 5g n-butyric acid-1-( 13 COOH) was dissolved in 50ml of benzene, 32.10g of oxalyl chloride was added at -20°C, and the reaction was stirred at 100°C for 2h until no HCl gas was released. The reaction liquid spins off the solvent to obtain the intermediate intermediate (1) n-butyryl chloride- 13 C5.74g.
[0045] 5.74g intermediate (1) n-butyryl chloride- 13 Dissolve C in chloroform, heat to 60°C, slowly add 20.50 g of dry liquid bromine dropwise under stirring, and continue stirring for 2 hours. After the reaction, the reactant was cooled to room temperature. Spin to remove the solvent, distill under reduced pressure, collect 150 ° C light yellow oily fraction intermediate (2) 2-bromobutyric acid chloride- 13 C8.91g.
[0046] Dissolve 15.50g of urotropine in 100ml of isopropanol, stir and mix. Dry ammonia gas was introduced at room temperature until the pH of the soluti...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More