

Tumor marker screening method based on nucleosome distribution characteristics and application
A technology of tumor markers and screening methods, applied in the field of quantification of nucleosome distribution, can solve problems such as screening of tumor biomarkers
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

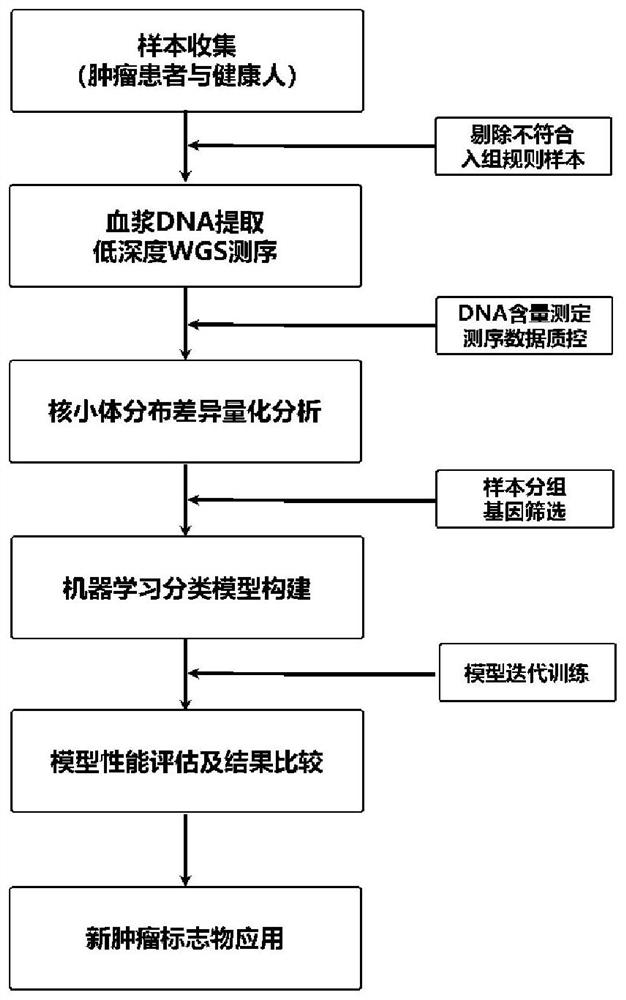
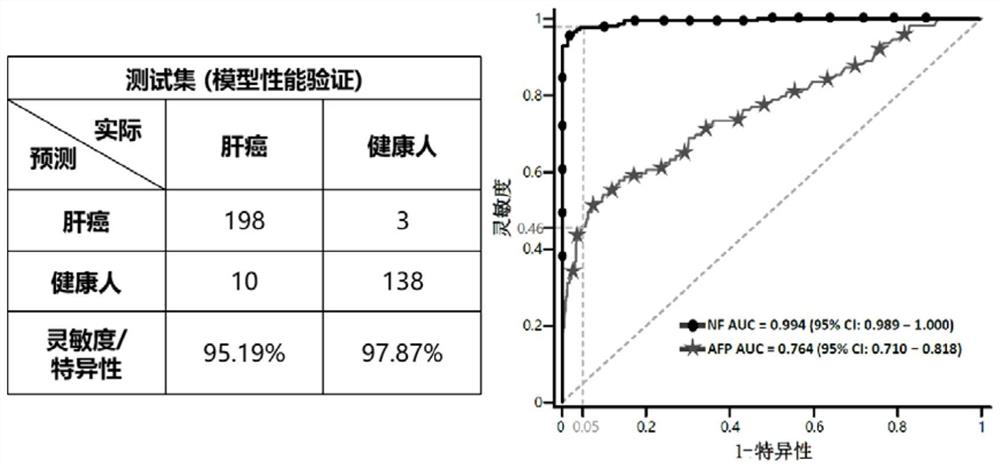
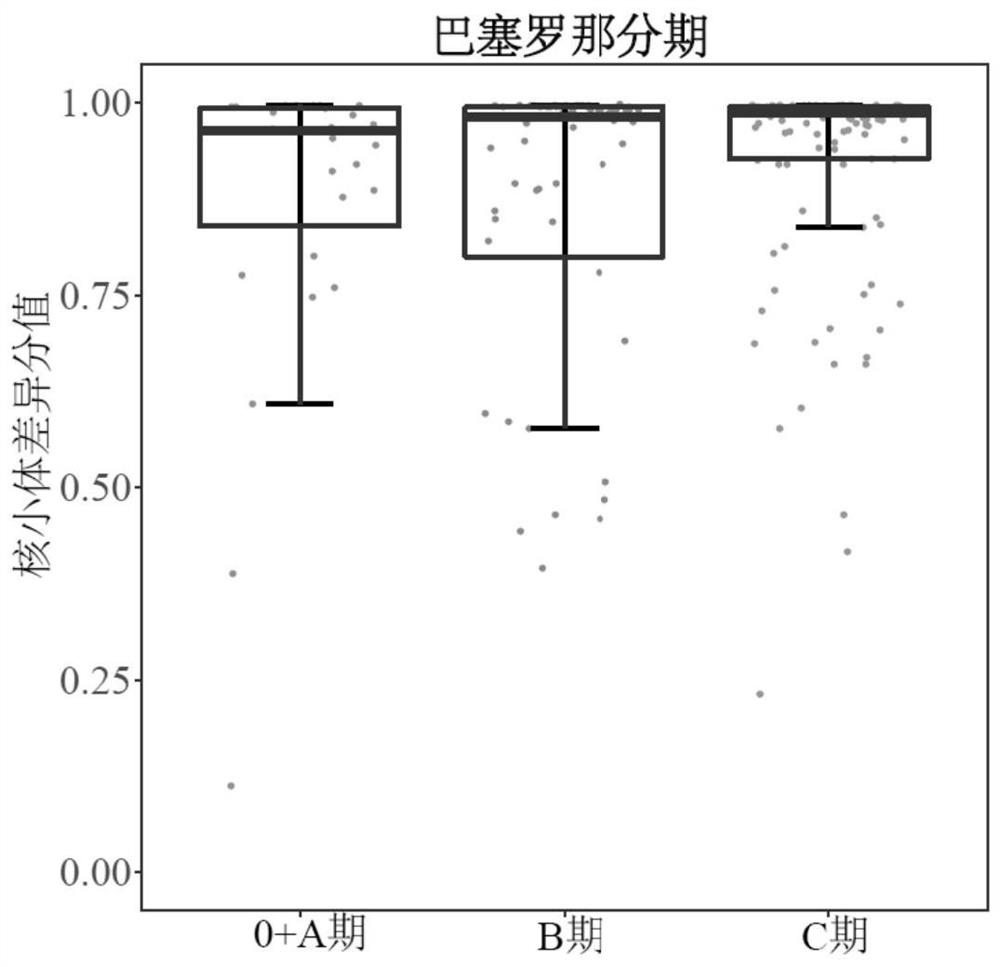
Examples
Embodiment 1
[0096] Determine the enrolled tumor patients and healthy people. A total of 984 subjects were recruited from 508 patients with liver cancer (HCC) and 476 healthy controls (NC) from two centers. According to the pathological diagnosis, 27 liver cancer samples were excluded, and 957 subjects were finally determined ( figure 1 ).
[0097] Plasma cell-free DNA (cfDNA) was extracted from tumor patients and healthy people. Take 3ml of peripheral blood from each participant (collected and stored in Streck cell-free DNA blood collection tubes), use Eppendorf centrifuges (5810R and 5427R, German), and centrifuge at 1600g at low speed for 10min at a low temperature of 4°C, and only take the supernatant liquid; then centrifuged at 16000 g for 10 min at high speed, and the supernatant was taken to obtain a plasma sample. The free DNA in the plasma was extracted with a kit MagMAX Cell-Free DNA Isolation Kit (Thermo) and a nucleic acid extractor (Thermo Kingfisher FLEX, USA). The concen...
Embodiment 2
[0099] Low-depth whole-genome sequencing was performed on the cfDNA samples of all participants prepared in Example 1. The sequencing process is as follows:
[0100] (1) WGS library construction and on-machine sequencing: take 5ng cfDNA and use the Enzymatics (USA) kit to construct a pre-library, mainly including two steps of end repair (5X ER / A-Tailing Enzyme Mix) and adapter (WGS Ligase) , the adapter sequence is suitable for the Illumina NovaSeq 6000 sequencing platform. The adapters were ligated and purified using XP magnetic beads (Agencourt AMPure XP beads, Beckman Coulter). The concentration value of WGS library was determined by qPCR (KAPA Library QuantKit, Roche), and the library size was determined by Fragment Analyzer (Agilent, USA). Afterwards, paired-end 150bp sequencing was performed on the Illumina NovaSeq 6000 sequencing platform, and the average data volume of a single sample was 2X that of the whole genome.
[0101] (2) Data quality control: Use Fastp soft...
Embodiment 3
[0103] The sequencing data obtained in Example 2 was used to quantify the difference in nucleosome distribution in the transcription start site region of the genome-wide gene, and the specific steps were as follows:
[0104] (1) Acquisition of the gene transcription start site region of the whole genome: use the transcription start site of the main transcript of the reference gene published in the USCS database, and extend 2500 bp before and after each as the transcription start site region of the gene.
[0105] (2) Acquisition of the sequencing depth of the transcription start site region: divide the transcription start site region into 500 small regions, and use the average sequencing depth of each small region to represent the sequencing depth of the small region, thereby eliminating the sequencing depth Insufficient results in some sites not being covered by DNA fragments.
[0106](3) Delineation of the central area of the transcription start site, the edge area and the ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More