

Cleavage of nucleic acids
a nucleic acid and cleavage technology, applied in the field of nucleic acid cleavage, detection and characterization of nucleic acid sequences and sequence changes, can solve the problems of ineffective thermostable dna ligases, limited use of lcr for mutant screening, and inability to raise reaction temperatures to prevent, etc., to achieve rapid detection and identification of variants
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

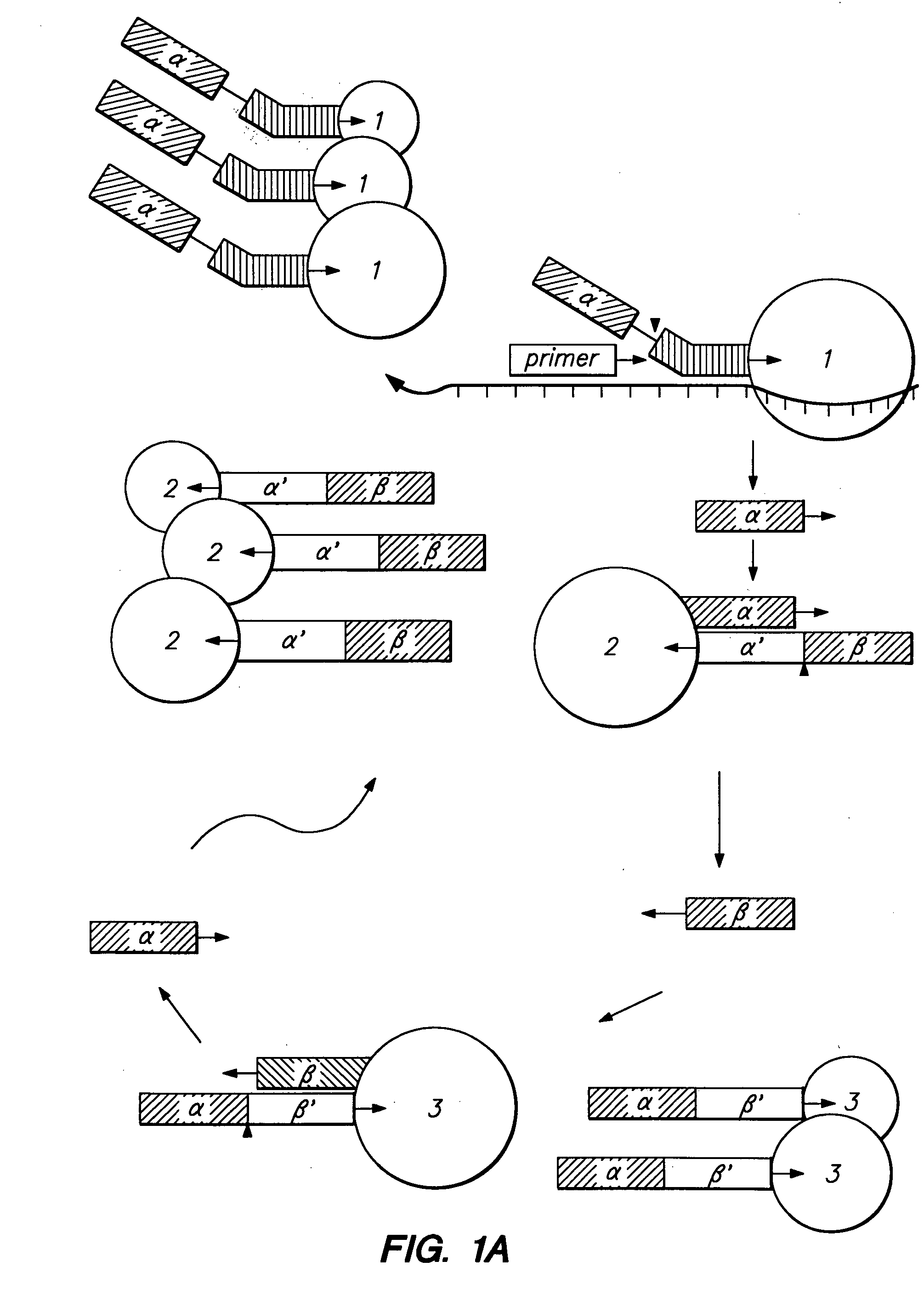
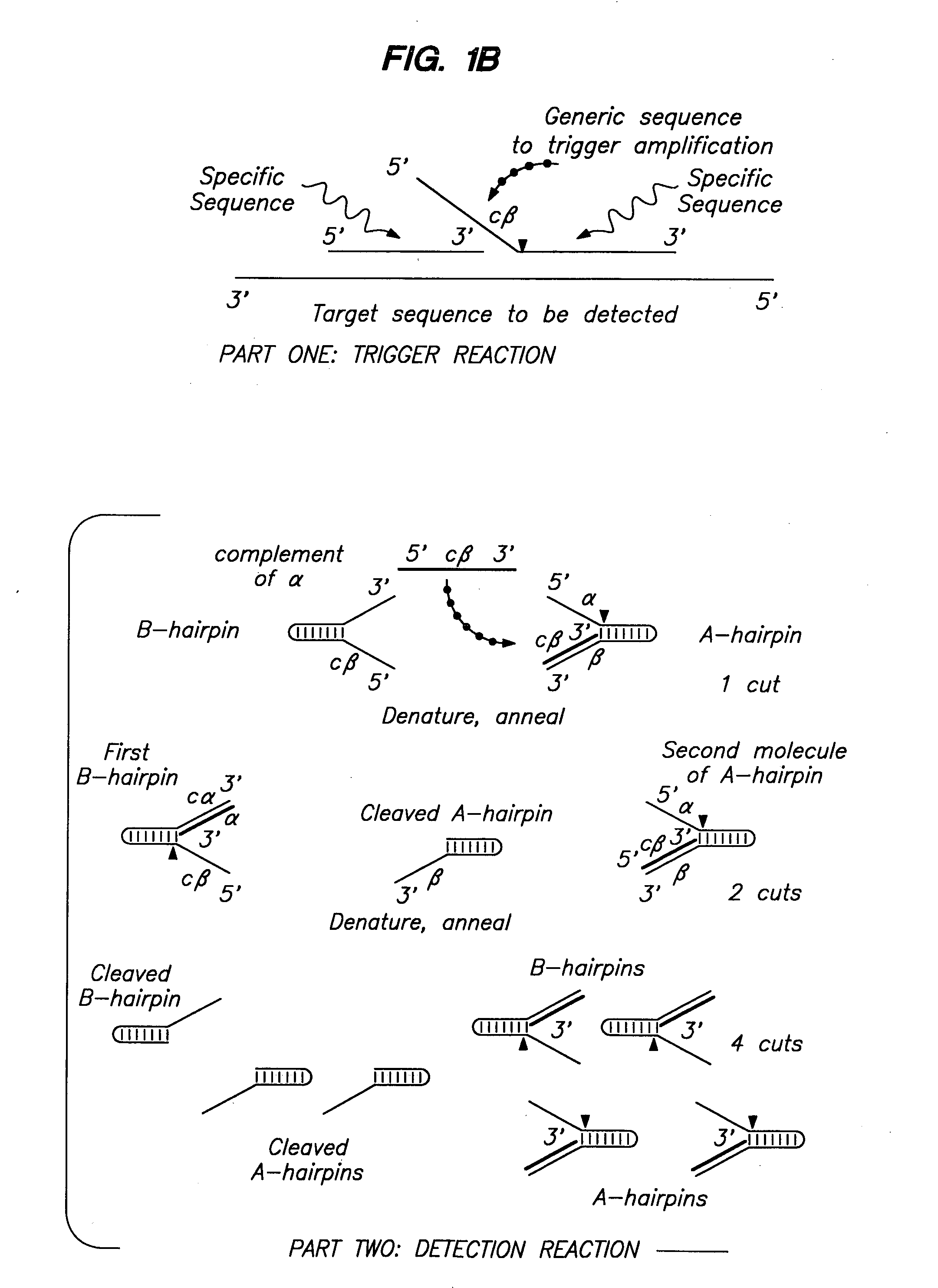

Examples
example 1
Characteristics of Native Thermostable DNA Polymerases
[0391] A. 5′ Nuclease Activity pf DNAPTaq
[0392] During the polymerase chain reaction (PCR) (Saiki et al., Science 239:487 (1988); Mullis and Faloona, Methods in Enzymology 155:335 (1987)), DNAPTaq is able to amplify many, but not all, DNA sequences. One sequence that cannot be amplified using DNAPTaq is shown in FIG. 6 (Hairpin structure is SEQ ID NO:15, PRIMERS are SEQ ID NOS:16-17.) This DNA sequence has the distinguishing characteristic of being able to fold on itself to form a hairpin with two single-stranded arms, which correspond to the primers used in PCR.
[0393] To test whether this failure to amplify is due to the 5′ nuclease activity of the enzyme, we compared the abilities of DNAPTaq and DNAPStf to amplify this DNA sequence during 30 cycles of PCR. Synthetic oligonucleotides were obtained from The Biotechnology Center at the University of Wisconsin-Madison. The DNAPTaq and DNAPStf were from Perkin Elmer (i.e., AMPLIT...
example 2
Generation of 5′ Nucleases from Thermostable DNA Polymerases
[0426] Thermostable DNA polymerases were generated which have reduced synthetic activity, an activity that is an undesirable side-reaction during DNA cleavage in the detection assay of the invention, yet have maintained thermostable nuclease activity. The result is a thermostable polymerase which cleaves nucleic acids DNA with extreme specificity.
[0427] Type A DNA polymerases from eubacteria of the genus Thermus share extensive protein sequence identity (90% in the polymerization domain, using the Lipman-Pearson method in the DNA analysis software from DNAStar, WI) and behave similarly in both polymerization and nuclease assays. Therefore, we have used the genes for the DNA polymerase of Thermus aquaticus (DNAPTaq) and Thermus flavus (DNAPTfl) as representatives of this class. Polymerase genes from other eubacterial organisms, such as Thermus thermophilus, Thermus sp., Thermotoga maritima, Thermosipho africanus and Bacill...
example 3
5′ Nucleases Derived from Thermostable DNA Polymerases can Cleave Short Hairpin Structures with Specificity
[0489] The ability of the 5′ nucleases to cleave hairpin structures to generate a cleaved hairpin structure suitable as a detection molecule was examined. The structure and sequence of the hairpin test molecule is shown in FIG. 19A (SEQ ID NO:15). The oligonucleotide (labeled “primer” in FIG. 19A, SEQ ID NO:22) is shown annealed to its complementary sequence on the 3′ arm of the hairpin test molecule. The hairpin test molecule was single-end labeled with 32P using a labeled T7 promoter primer in a polymerase chain reaction. The label is present on the 5′ arm of the hairpin test molecule and is represented by the star in FIG. 19A.
[0490] The cleavage reaction was performed by adding 10 fmoles of heat-denatured, end-labeled hairpin test molecule, 0.2 uM of the primer oligonucleotide (complementary to the 3′ arm of the hairpin), 50 μM of each dNTP and 0.5 units of DNAPTaq (Perkin...
PUM
| Property | Measurement | Unit |
|---|---|---|
| nucleic acid | aaaaa | aaaaa |
| fluorescent | aaaaa | aaaaa |
| nucleic acid cleavage structure | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More