New method for synthesizing besifloxacin
A technology of besifloxacin and synthetic method, applied in the field of preparation of besifloxacin, can solve the problems of difficult nucleophilic substitution, harsh reaction conditions, long reaction time, etc., and reduce the process and reaction conditions of chiral resolution Gentle, easy-to-go effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

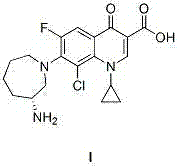
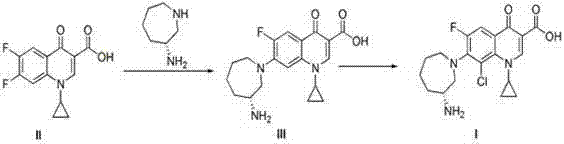
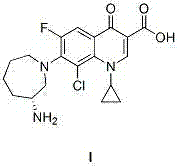
Examples
Embodiment 1
[0017] Example 1 L-lysine methyl ester (VI).
[0018] Dissolve L-lysine (1.46g, 0.01mol) in methanol (20ml), add thionyl chloride dropwise to the reaction solution at room temperature, after the dropwise addition, react at room temperature for 2 hours, TLC detection After the reaction was completed, the reaction solution was concentrated to obtain the crude compound VI (1.5 g, yield 84.4%).
Embodiment 2
[0019] Example 2 L-type aminocaprolactam (VII).
[0020] Dissolve L-lysine methyl ester (1.6g, 0.01mol) in methanol (40ml), stir well so that the solid material is fully dissolved, then add (1.08g, 0.02mol) sodium methoxide to the reaction solution, at room temperature Stir evenly, react at room temperature for 5 hours, then raise the temperature to 80°C and react for 12 hours, then cool down to 0°C, a white solid precipitates, filters, and washes with a little methanol to obtain the crude product of compound VII (0.98g, yield 76.6%).
Embodiment 3
[0021] Example 3 (R)-7-(3-aminohexahydro-1H-azepine-1-yl)-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3quine Phinolic acid (III).
[0022] Dissolve intermediate II (2.83g, 0.01mol) in acetonitrile (30ml), stir until intermediate II is completely dissolved, then slowly add side chain L-type aminocaprolactam (2.58g, 0.02mol) dropwise to the reaction solution, After the dropwise addition is complete, add triethylamine (3.03g, 0.03mol) to the reaction solution, stir well, heat and reflux for 10 hours, after TLC detects that the reaction is complete, concentrate the reaction solution, add 80ml of dichloromethane, and saturate with NaCl Solution (50ml*3) washed the organic phase, dried the organic phase with anhydrous sodium sulfate, and concentrated the reaction solution to obtain the crude compound III (2.8 g, yield 77.9%).
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com