Preparation method of sofosbuvir intermediate
A technology for intermediates and preparation steps, which is applied in the field of preparation of sofosbuvir intermediates, can solve problems such as cumbersome operations, high waste treatment costs, and high environmental protection pressure, and achieve high stereoselectivity, novel synthetic routes, and high yields. high effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

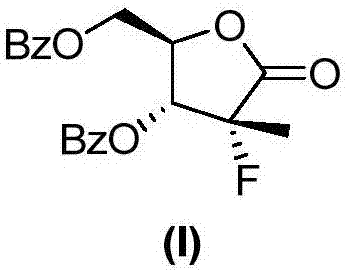
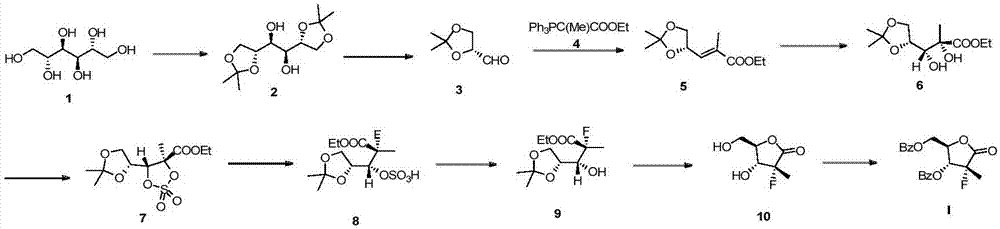
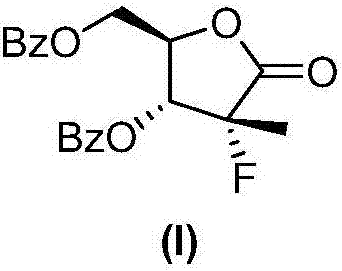
Examples
Embodiment 1
[0070] Under the protection of nitrogen, the compound of formula II (50g, 0.31mol) was dissolved in dry tetrahydrofuran (1000mL), cooled to -50°C, and n-butyllithium (2.5M, 135mL, 0.34mol) was slowly added dropwise, controlled The inner temperature is not higher than -40 degrees. After dropping, stir at about -50°C for 30 minutes. Then the compound of formula III (72.9 g, 0.34 mol) was added dropwise, the internal temperature was controlled not to be higher than -40°C, and after the addition was completed, it was stirred at about -50°C for 1 hour. After the reaction was completed, it was quenched with saturated aqueous ammonium chloride (200 mL), and extracted twice with ethyl acetate (500 mL). The organic phases were combined, dried over anhydrous sodium sulfate, and concentrated. The obtained crude product was slurried by adding n-hexane (500 mL), filtered and dried to obtain the compound of formula IV.
[0071] Yield: 75.8 g, Yield: 83%.
Embodiment 2
[0073] Under the protection of nitrogen, the compound of formula II (50g, 0.31mol) was dissolved in dry tetrahydrofuran (1000mL), triethylamine (154.9g, 1.53mol), DMAP (3.7g, 0.031mol) were added, cooled to 0- At 5°C, the compound of formula III (99.3 g, 0.46 mol) was added dropwise. After the addition was complete, the mixture was raised to room temperature and reacted overnight. Quenched with saturated aqueous ammonium chloride (200 mL), extracted twice with ethyl acetate (500 mL). The organic phases were combined, dried over anhydrous sodium sulfate, and concentrated. The resulting crude product was subjected to column chromatography to obtain a compound of formula IV.
[0074] Yield: 71.3 g, Yield: 78%.
Embodiment 3
[0076] The compound of formula II (250 g, 1.53 mol) was added to toluene (3750 mL), and heated to 70 degrees. Sodium methoxide in methanol (29 wt%, 314 g, 1.68 mol) was added dropwise. After the addition, methanol was distilled off under normal pressure. Cool to 0-5°C, add the compound of formula III (364.4 g, 1.68 mol) dropwise, and control the temperature not higher than 20°C. After adding, react for 1-2 hours. After the reaction was completed, water (250 mL) was added, heated to 50-60° C., the layers were separated, and the water phase was separated. The organic phase was washed twice with water. The organic phase was concentrated to give crude product. Add n-hexane (1500mL) to the crude product to make a slurry, filter and dry to obtain the intermediate of formula IV.
[0077] Yield: 436 g, Yield: 95.4%.
[0078] Among the above-mentioned examples 1-3, the base n-BuLi or triethylamine or sodium methylate used can also be selected from iso-BuLi, t-BuLi, LDA, LiHMDS, K...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com