

Preparing method for medicament midbody for treating cystic fibrosis
An intermediate and nitration reaction technology, applied in the preparation of nitro compounds, organic chemistry, etc., can solve the problems of difficult removal, complex nitration reaction products, large solvent and silica gel consumption in column chromatography, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

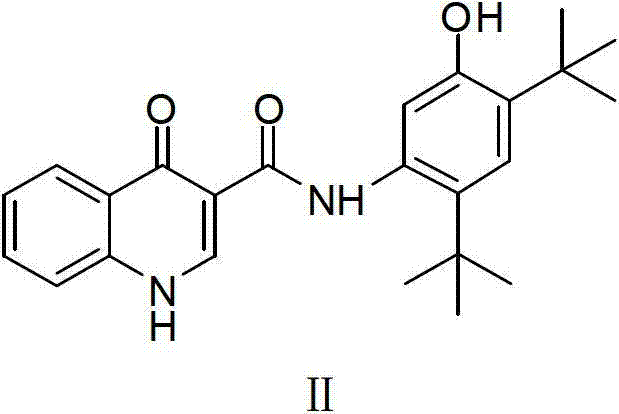
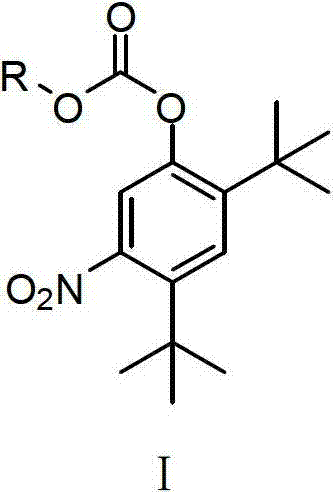
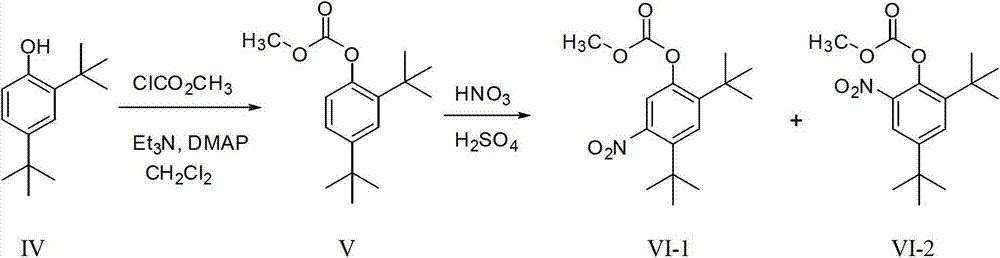
Examples
Embodiment 1
[0039] 2,4-di-tert-butylphenyl ethyl carbonate (III, R=CH 2 CH 3 )Synthesis
[0040] Add compound IV (50g, 242.3mmol) and 600ml ethyl acetate into a 1L three-necked flask, stir and dissolve at room temperature, add triethylamine (55ml, 394.6mmol) after dissolution, cool to 0°C in an ice bath, and add dropwise Ethyl chloroformate (50ml, 525.2mmol), after adding, return to room temperature and react for 2~4h, filter with suction, wash the filtrate with water and saturated sodium chloride solution successively, separate the organic layer, anhydrous NaSO 4 After drying, filtering, and concentrating, 66.7 g of crude 2,4-di-tert-butylphenyl ethyl carbonate was obtained, with a yield of 98.9%, in the form of light yellow oil.
[0041] 1 H-NMR (300MHz, CDCL 3 ), δ(ppm): 7.38 (1H, d, J=2.3Hz, ArH), 7.24 (1H, dd, J 1 =8.5Hz,J 2 =2.4Hz, ArH), 7.0 (1H, d, J=8.4Hz, ArH), 4.32 (2H, q, J=7.1Hz, CH 2 ), 1.41 (9H, s, CH 3 ), 1.36 (3H, t, J=7.1Hz, CH 3 ), 1.29 (9H, s, CH 3 ).
[004...
Embodiment 2
[0046] 2,4-Di-tert-butyl-5-amino-phenylcarbonate ethyl ester (IX, R=CH 2 CH 3 )Synthesis
[0047]Put compound I (20g, 61.8mmol) into a 500ml single-necked bottle, add 250ml of anhydrous methanol, stir to dissolve, add 2g of 10% Pd / C, and react at room temperature for 2 to 4 hours in a hydrogen atmosphere, then remove palladium carbon by suction filtration, Concentrate the filtrate to remove methanol until solids appear in the bottle, stop the concentration, refrigerate overnight in the refrigerator, filter with suction, wash the filter cake with a small amount of methanol to obtain bright gray crystals, and repeat the above operation for the mother liquor to obtain 10.3 g of bright gray crystals, with a yield of 56.8%. m.p.142-144°C.
[0048] 1 H-NMR (300MHz, CDCL 3 ), δ(ppm): 7.20 (1H, s, ArH), 6.38 (1H, s, ArH), 4.32 (2H, q, J=7.1Hz, CH 2 ), 1.52 (3H, t, J=7.1Hz, CH 3 ), 1.42 (9H, s, CH 3 ), 1.35 (9H, s, CH 3 ). Carbonic acid 2,4-di-tert-butyl-5-(1,4-dihydro-4-oxoq...
Embodiment 3
[0055] Carbonic acid 2,4-di-tert-butyl-5-nitro-phenyl ethyl ester (I, R=CH 2 CH 3 )Synthesis
[0056] Add compound III (20g, 71.8mmol) and 70ml of dichloromethane into a 250ml three-necked flask, cool in an ice bath to 0-5°C, add concentrated sulfuric acid (16ml) dropwise under stirring, dropwise for about 0.5 hours, then drop Add concentrated nitric acid (8ml), drop it, stir and react at 0-5°C for 0.5 hours, pour the reaction solution into 250ml of ice water, separate the organic layer, extract the aqueous layer with dichloromethane, combine the organic layers, and successively water, Wash with saturated NaCl solution, anhydrous NaSO 4 Dry and concentrate to obtain a yellow oil, add 5ml of petroleum ether, stir for 1 minute, refrigerate and crystallize overnight, a large number of light yellow crystals precipitate, filter with suction, wash the filter cake with petroleum ether until it is nearly white, after the mother liquor is concentrated, add 2.5ml of petroleum ether ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More