

Method for preparing solithromycin
A technology for solithromycin and intermediates, which is applied in the field of preparing solithromycin and can solve the problems of difficulty in producing solithromycin, poor solubility, low conversion rate and the like
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

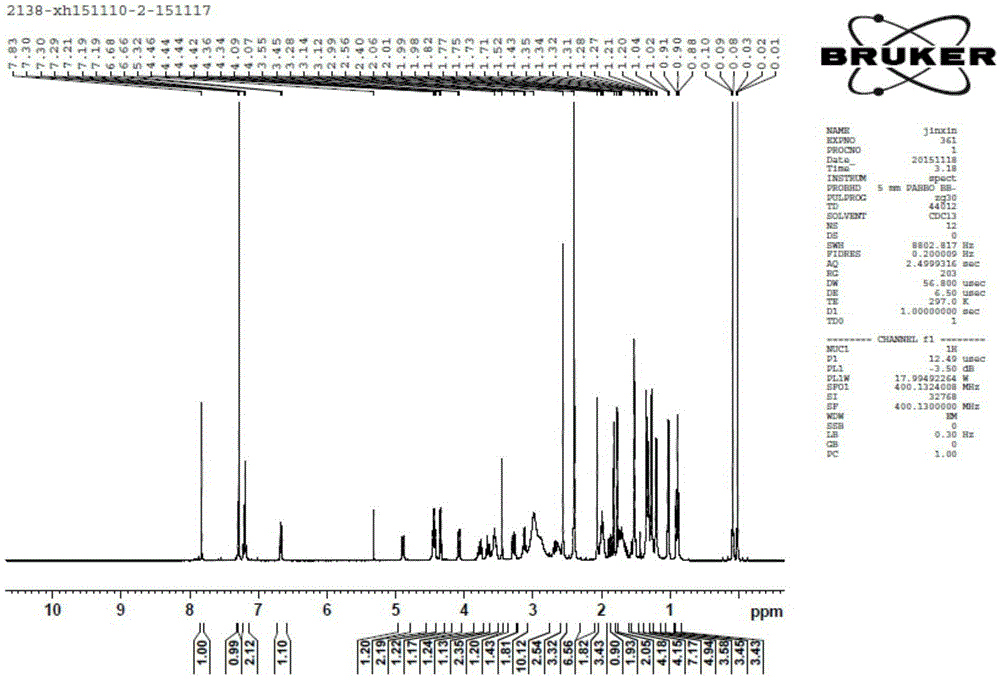

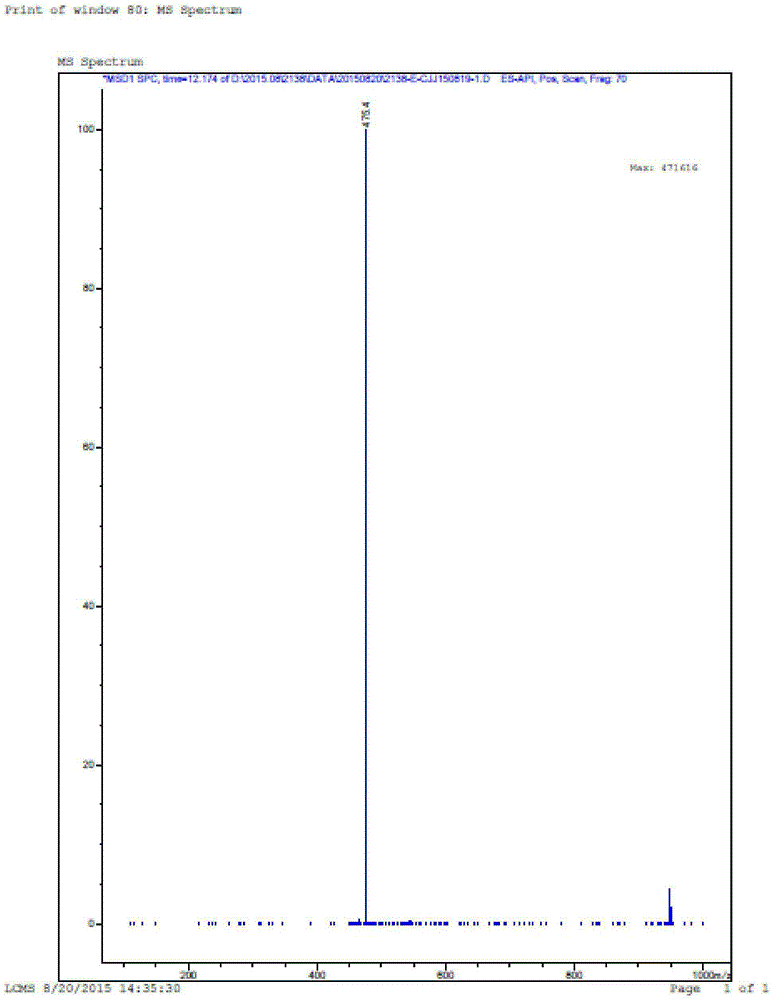
Examples
Embodiment 1
[0094] Preparation of Intermediate II:
[0095]
[0096] Add clarithromycin (50 g, 0.067 mol), triethylamine (18.75 mL, 0.135 mol, 2 equivalents), and ethyl acetate (350 mL) into a 500 mL reaction flask and stir to mix. Benzoic anhydride (22.5 g, 0.1 mol, 1.5 equiv) was added in portions. After the addition, stir at room temperature (20-25°C) for 24h. After detecting that the clarithromycin reaction was complete, the solvent was distilled off under reduced pressure (temperature<45° C.). Add 500 mL ice methanol to the residue, stir in ice bath (0-5° C.) for 0.5 h, and filter with suction. The filter cake was rinsed with ice methanol (100 mL×2) and dried in vacuo to obtain 56 g of white solid.
[0097] ESI[M+1]:852
[0098]
[0099] Add intermediate A (56 g, 0.0657 mol), ethanol (300 mL) and water (300 mL) obtained in the previous step reaction into a 1000 mL reaction flask and mix and stir. Concentrated hydrochloric acid (56 mL, 0.672 mol, 10 equivalents) was slowly ...
Embodiment 2
[0124] Preparation of Oxazole Ring Intermediate IIIa
[0125]
[0126] Add compound II (5.7g, 0.0075mol), azidobutylamine (1.2g, 0.0105mol, 2 equivalents), acetonitrile (40mL), 1,8-diazabicycloundecane-7 in a 250mL reaction flask -ene DBU (0.5 mL), react overnight at 55-65°C. After the reaction of Compound II was detected by sampling, the solvent was evaporated under reduced pressure (suspension evaporation, less than 0.01MPa, 40-45°C), the residue was diluted with water (50mL), extracted with ethyl acetate (50mL×2), and the organic phases were combined. Wash with saturated brine (30mL), dry over anhydrous sodium sulfate, filter with suction, spin dry (suspension evaporation, less than 0.01MPa, 40-45°C), and purify by column chromatography (25×500mm silica gel column, silica gel 200-300 The eluent was dichloromethane:methanol=10:1), and 4g of white solid was obtained, which was Intermediate IIIa.
[0127] ESI[M+1]:814.
Embodiment 3
[0129] Preparation of intermediate IV by fluorination reaction
[0130]
[0131] Add intermediate IIIa (1.63 g, 0.002 mol) and tetrahydrofuran (20 mL) into a 100 mL three-neck reaction flask, replace with nitrogen, and stir to dissolve. The reaction solution was cooled to -30°C, and potassium tert-butoxide (0.45 g, 0.005 mol, 2.5 equivalents) was added in portions. After the addition, keep stirring at -20~-25°C for 1h. N-fluorobisbenzenesulfonamide NFSI (1 g, 0.003 mol, 1.5 equiv) was added in portions. After the addition, keep stirring at -20~-25°C for 1.5h. After the reaction of intermediate IIIa was detected by sampling HPLC, 5% aqueous sodium bicarbonate (50 mL) was added to quench the reaction, extracted with ethyl acetate (100 mL), washed with saturated brine (30 mL), dried over anhydrous sodium sulfate, suction filtered, and spun After drying, 1.36 g of light yellow solid was obtained, which was Intermediate IV, which could be directly used in the next reaction wi...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More