62:676-689; Gebhart et al., 1998, Int. J. Oncol. 12:1151-1155; Hacia et al., 1996, Nat. Genet. 14:441-447, all of which are incorporated herein by reference), this one-by-one query is an inefficient and incomplete method for genetically
typing cells.
This method is very likely to yield useful information about
cancer, but suffers limitations.
First, the interpretation of the data obtained and its correlation with
disease process is likely to be a complex and difficult problem: multiple changes in
gene expression will be observed that are not relevant to the
disease of interest.
Second, our present cDNA collections are not complete, and any
chip is likely to be obsolete in the near future.
Third, while a picture of the current state of the
cell might be obtained, there would be little direct information about how the
cell arrived at that state.
Lastly, obtaining reliable mRNA from biopsies is likely to be a difficult problem, because
RNA is very unstable and undergoes rapid degradation due to the presence of ubiquitous RNAses.
Hence, in its unaltered format, the simple DNA probe
chip would not suffice for the robust detection of genomic sequences.
Because each address contains fragments derived from the entire
BAC clone, several problems are created.
Also, the great size of the megacloning vector inserts limits the positional resolution.
Another drawback is the presence of DNA derived from the megacloning vector and host sequences.
The steps of excising and purifying the
genomic DNA inserts from the vector and host sequences complicate and hinder rapid fabrication of microarrays.
Analysis of the genetic changes in human tumors is often problematic because of the presence of normal stroma.
Samples of
tumor tissue are often contaminated with non-cancerous cells, making isolation and study of tumor
cell DNA difficult.
While either
microdissection or
flow cytometry can produce small samples highly enriched for
tumor cells or nuclei, the amount of extracted DNA recoverable from such enriched samples is insufficient for most uses.
One limitation of the
amplicon useful in RDA is that an
amplicon representation with much lower complexity than that of the
genome from which the
amplicon is derived is needed to enable the subtractive hybridization to proceed effectively.
Such
low complexity representations (LCRs) do not "capture" enough (typically, 7% or less) of the
genome to be generally useful for other applications.
The complexity of the representation is related to the frequency of
cutting of the
restriction enzyme used to generate the genomic fragments, combined with the amplification reaction steps, e.g., PCR, which tend to favor the smaller fragments.
Therefore the amplification is not reliably reproducible.
This makes the use of such whole genomic amplifications for the purposes of sample to sample comparisons difficult.
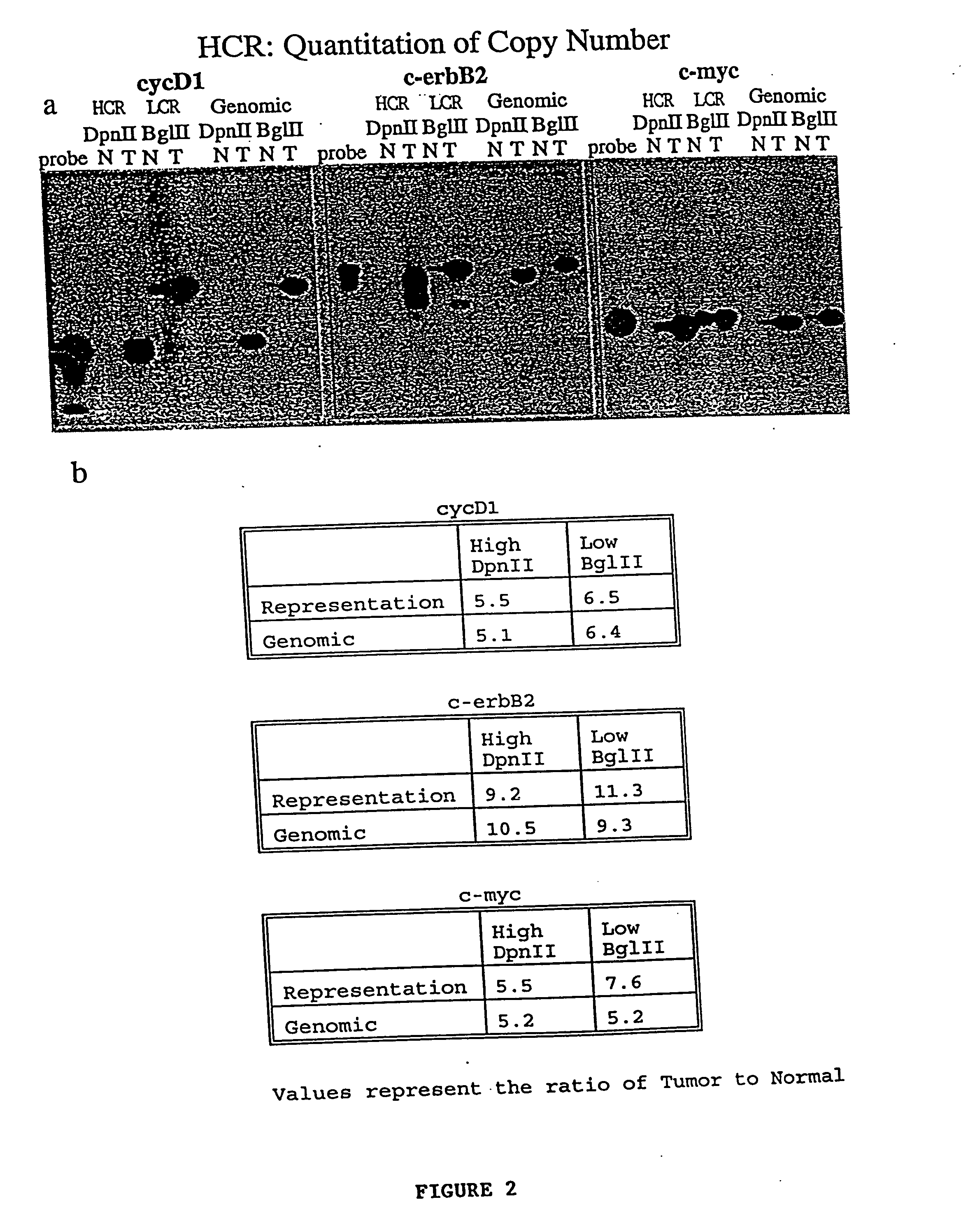
3. Whole genomic amplifications are not useful for quantitating the copy number of genes present in the original sample.
Thus, the abundance of each
gene relative to other genes in the original sample is not preserved during the amplification, making quantitation of copy number impossible.





 Login to View More
Login to View More