Kit and method for detecting vibrio parahaemolyticus in aquatic product
A technology for vibrio hemolyticus and aquatic products, which is applied in the fields of biochemical equipment and methods, microbe measurement/inspection, and resistance to vector-borne diseases, etc. It can solve the problems of high false negative rate of frozen food, low bacterial sensitivity, and long culture period To achieve the effect of improving food safety level, simple operation steps and short detection cycle
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

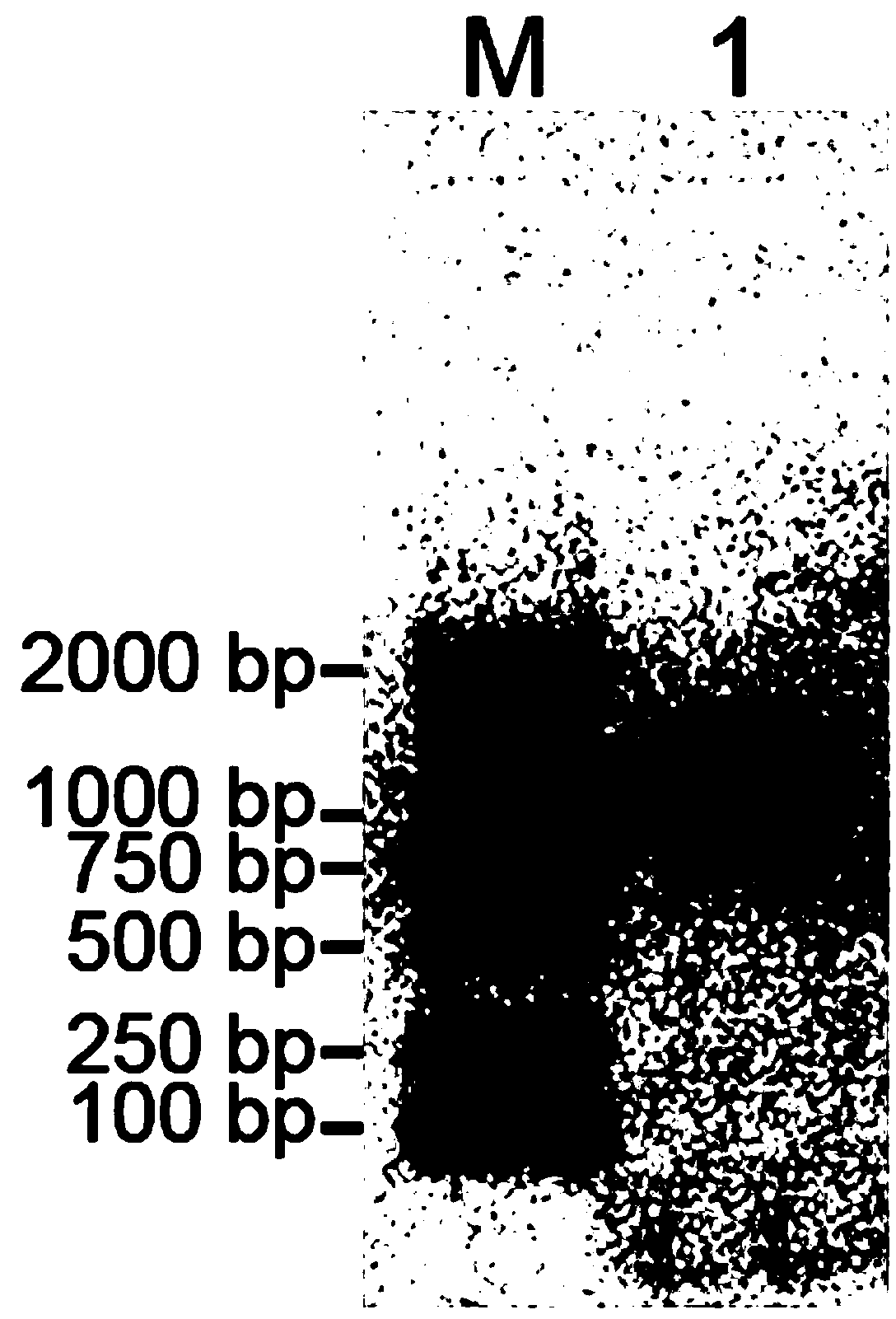
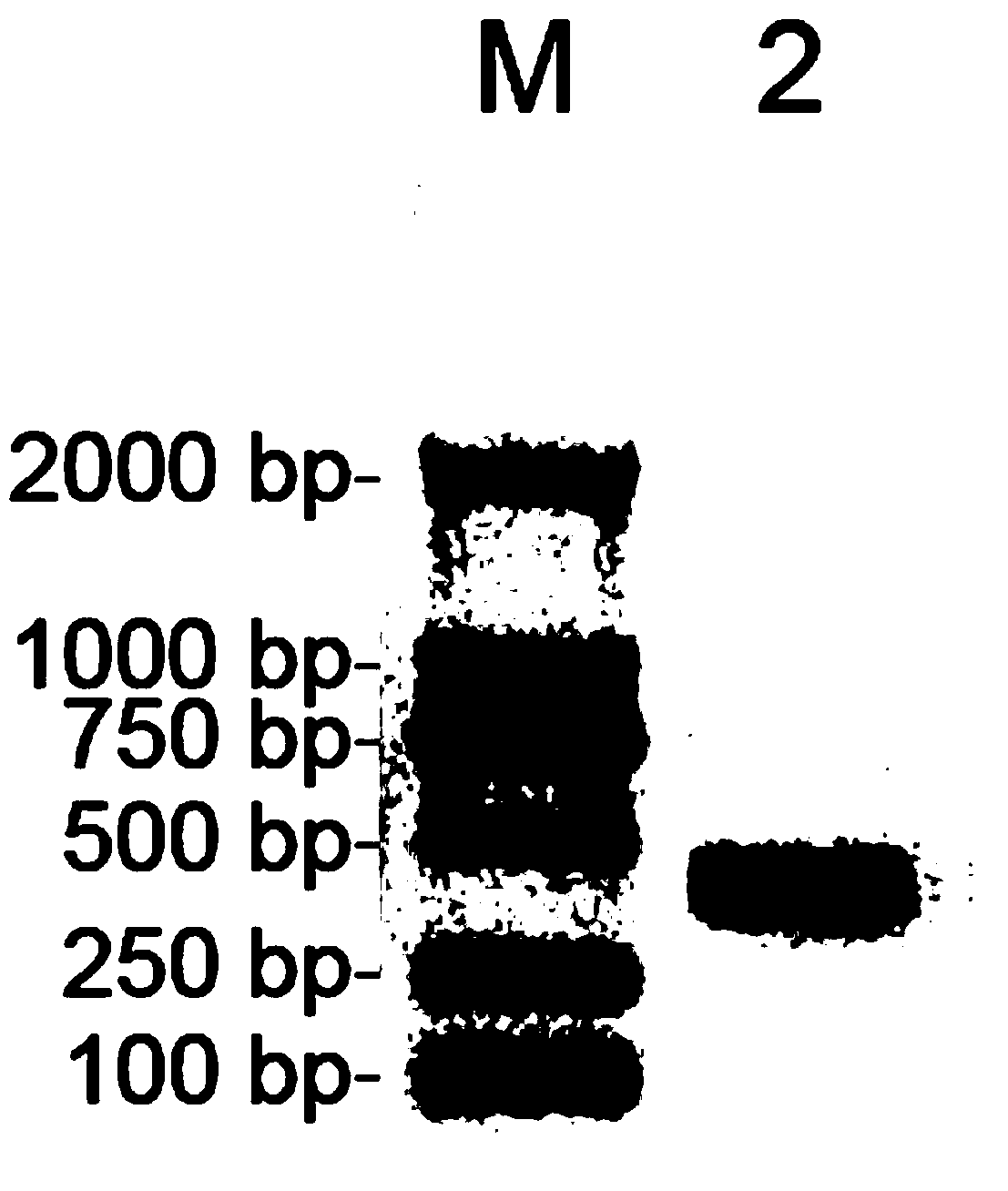
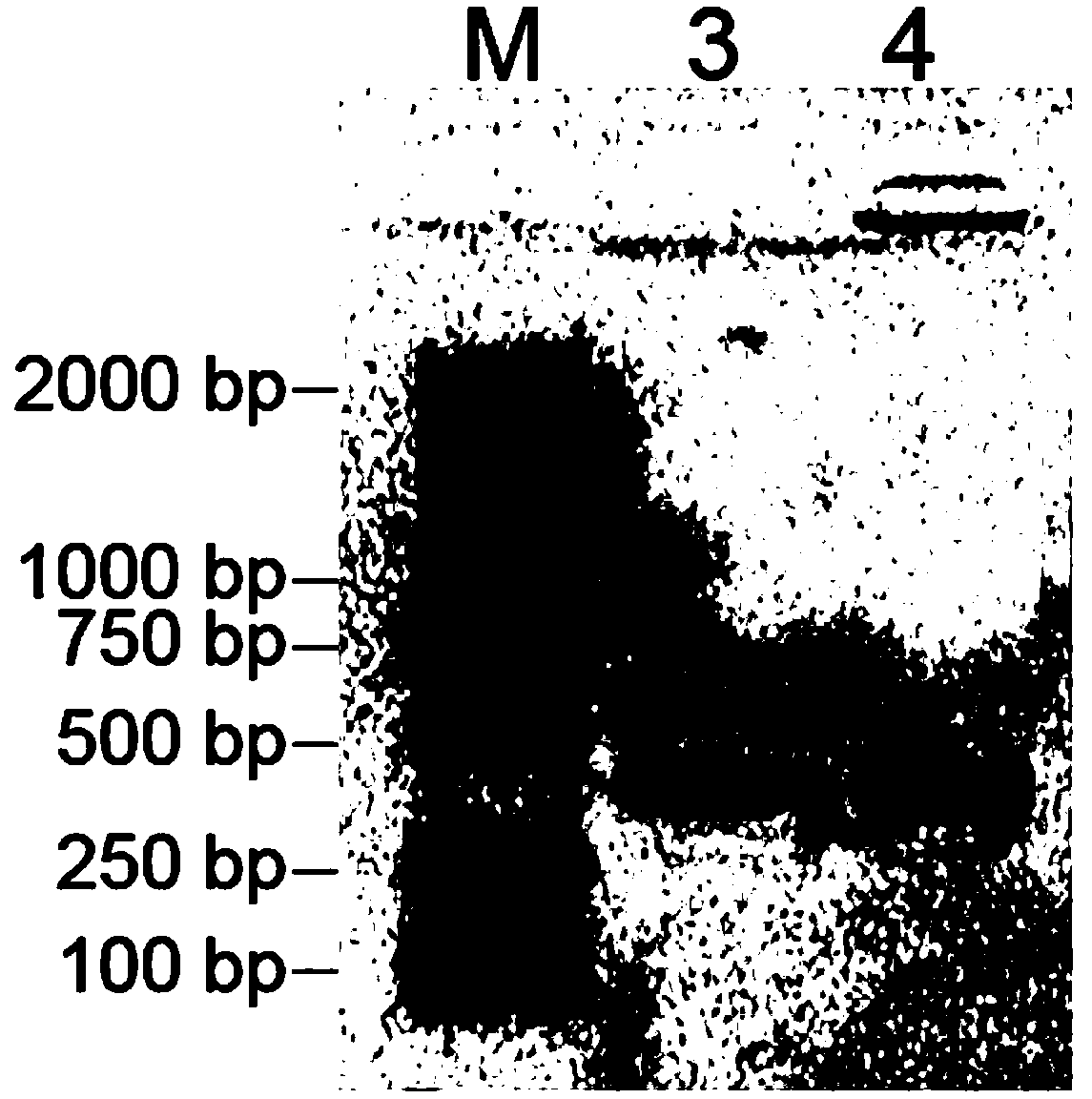
Examples
Embodiment
[0040] A kit for detecting Vibrio parahaemolyticus in aquatic products, including sterile deionized water, PCR reaction solution, Taq DNA polymerase, ethidium bromide DNA dye, bromophenol blue loading buffer, standard, control;
[0041] Wherein, the PCR reaction liquid contains PCR reaction buffer, MgCl 2 , deoxyribonucleoside triphosphates (dNTPs), primers for detection;
[0042] The detection primers were the mixture of the following 4 primers (Table 1).
[0043] Table 1
[0044]
[0045] The primers used for detection are the D-type amino acid dehydrogenase small subunit gene of Vibrio parahaemolyticus as the template DNA, designed using Primer Premier 5, and synthesized by Sangon Bioengineering (Shanghai) Co., Ltd.;
[0046] The standard product is a recombinant plasmid containing the amplified product obtained after PCR amplification using the Vibrio parahaemolyticus genome as a template DNA and using P3 and P4 as primers. It is constructed by the following method:...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - Generate Ideas
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com