Although short molecules can be analyzed, considerable effort is required to assemble the information from the analysis of the short molecules into a description of the larger piece of DNA.
The major disadvantages of the
PCR method to amplify DNA are that 1) information about two flanking sequences must be known in order to specify the sequences of the primers; 2) synthesis of primers is expensive; 3) the level of amplification achieved depends strongly on the primer sequences, source DNA sequence, and the molecular weight of the amplified DNA; and 4) the length of amplified DNA is usually limited to less than 5 kb, although “long-distance” PCR (Cheng, 1994) allows molecules as long as 20 kb to be amplified.
The disadvantages of all “one-sided PCR” methods is that a) the length of the products are restricted by the limitation of PCR (normally about 2 kb, but with special reagents up to 50 kb); b) whenever the products are single DNA molecules longer than 1 kb they are too long to directly sequence; c) in
ligation-mediated PCR the
amplicon lengths are very unpredictable due to random distances between the universal priming site and the specific priming site(s), resulting in some products that are sometimes too short to walk significant distance, some which are preferentially amplified due to small size, and some that are too long to amplify and analyze; and d) in methods that use terminal
transferase to add a
polynucleotide tail to the end of a primer extention product, there is great heterogeneity in the length of the amplicons due to sequence-dependent differences in the rate of
primer extension.
The extent of the strand displacement reaction is not controlled and therefore the lengths of the product strands are not uniform.
The length of the product is very large—typically too large to be directly sequenced.
These steps, especially the DNA isolation step, are costly and
time consuming.
The
disadvantage of preparing DNA by amplifying random fragments of DNA is that considerable effort is necessary to assemble the information within the short fragments into a description of the original, source DNA molecule.
However, even after sequencing enough fragments that the average region has been sequenced 5-10 times, there will still be gaps between contigs due to statistical sampling effects and to systematic under-representation of some sequences during
cloning or PCR amplification (ref).
Thus the
disadvantage of sequencing random fragments of DNA is that 1) a 5-10 fold excess of DNA must be isolated, subjected to sequencing reactions, and analyzed before having large contiguous sequenced regions; and 2) there are still numerous gaps in the sequence that must be filled by expensive and time-consuming steps.
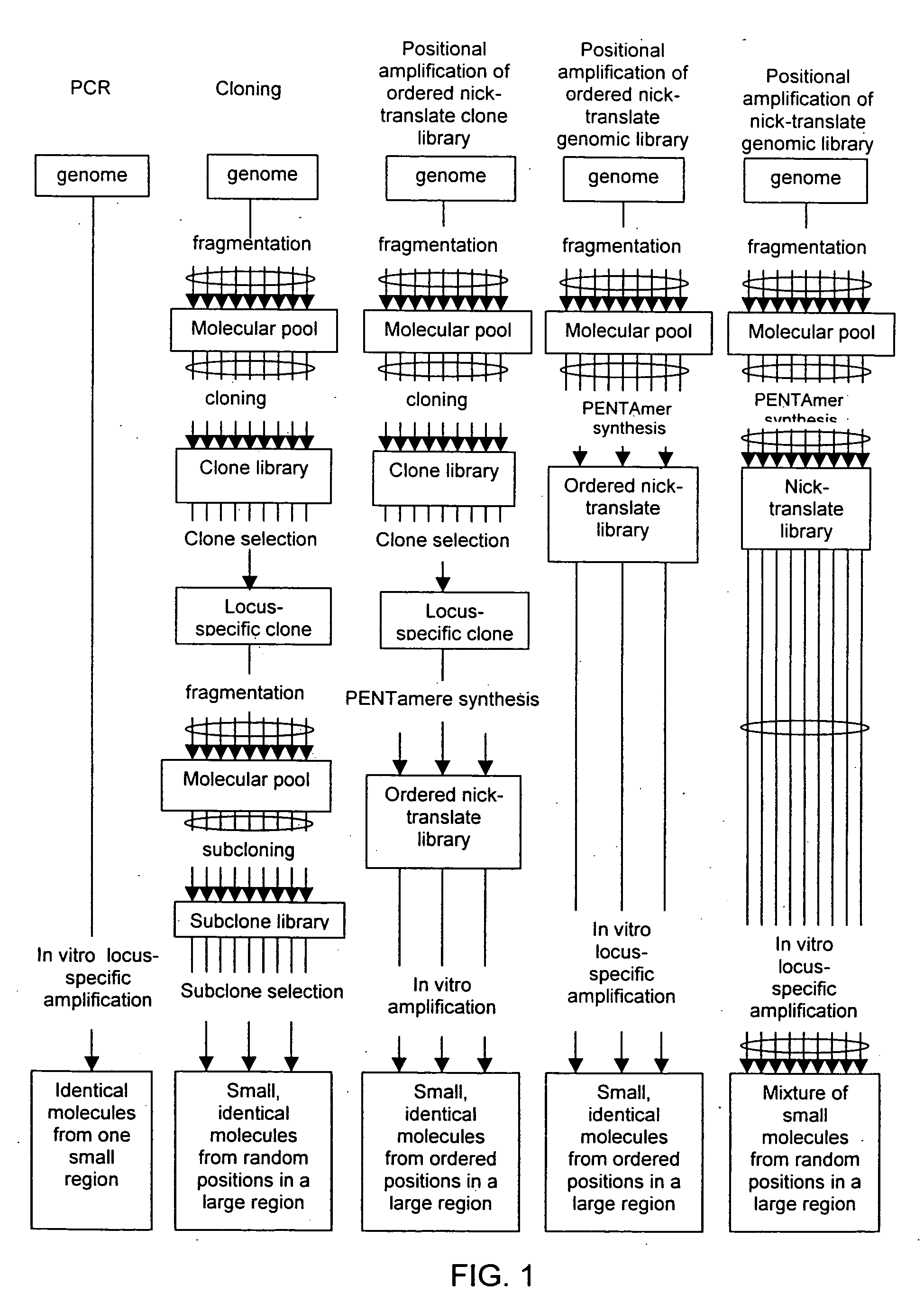
In vitro libraries are rarely used in
genomics, because the methods that exist for creating such libraries do not offer advantages over cloned libraries.
In particular, the methods used to amplify the
in vitro libraries are not able to amplify all the DNA in an unbiased manner, because of the size and sequence dependence of amplification efficiency.
The size separation and recloning steps make both of these methods labor intensive and slow.
They are generally limited to covering regions less than 10 kb in size and cannot be used directly on
genomic DNA but rather cloned DNA molecules.
No
in vivo methods are known are known to directly create ordered libraries of
genomic DNA.
This method required substantial effort to produce and order the PCR products for the job of sequencing cloned DNA.
No
in vitro methods are known to directly create ordered genomic libraries of DNA.
This method is useful for locating clones that overlap a common site (e.g., a specific
gene) in the
genome, but is too laborious to create an ordered
library of an entire
genome.
In addition many organisms have large amounts of repetitive DNA that can give false indications of overlap between two regions.
This method is insensitive to the presence of repetitive DNA.
Physical mapping of restriction sites is a very tedious process, because of the very large numbers of clones that have to be evaluated.
The Maxim-Gilbert method involves dangerous chemicals, and is time- and labor- intensive.
Commonly-used electrophoretic techniques for separating the dideoxyribonucleotide-terminated DNA molecules are limited to resolving sequencing ladders shorter than 500-1000 bases.
These methods have no substantial
advantage over the Sanger method.
It is difficult to amplify such short pieces of DNA for sequencing.
However, even if sequencing many random 50 bp pieces were possible, assembling the short, sometimes overlapping sequences into the
complete sequence of a large piece of DNA would be impossible.
The use of sequencing by hybridization is currently limited to resequencing, that is testing the sequence of regions that have already been sequenced.
Because it is currently very difficult to separate DNA molecules longer than.
This procedure is called “primer walking.” The requirement to synthesize a new
oligonucleotide every 400-1000 bp makes this method expensive.
The method is slow, because each step is done in series rather than in parallel.
In addition, each new primer has a significant
failure rate until optimum conditions are determined.
However the
exonuclease activity cannot be controlled to give a narrow distribution in molecular weights, so typically the
exonuclease-treated DNA is separated by
electrophoresis to better select the position of the end of the DNA samples before
cloning.
The labor-intense steps of clone screening make these methods impractical except for DNA less than about 10 kb long.
Although this method-was designed to walk along megabase distances along chromosomes, it was never put into practical use because of the necessity to maintain and screen hundreds of thousands of clones from each size fraction.
However, in the process of fragmentation all information about the relative positions of the fragment sequences in the native
genome is lost.
However, due to some regions being more difficult to clone than others and due to incomplete statistical sampling, there will still be some regions within the genome that are not sequenced even after highly redundant sequencing.
There are several disadvantages to the pure
shotgun strategy: 1) as the size of the region to be sequenced increases, the effort of assembling a contiguous sequence from
shotgun reads increases faster than N 1 nN, where N is the number of reads; 2) repetitive DNA and sequencing errors can cause ambiguities in
sequence assembly; and 3) because subclones from the entire genome are sequenced at the same time and significant redundancy of sequencing is necessary to get contigs of moderate size, about 50% of the sequencing has to be finished before the sequence accuracy and the
contig sizes are sufficient to get substantial information about the genome.
Focusing the sequencing effort on one region is impossible.
Sequencing random subclones is highly inefficient, because significant gaps exist until the subclones have been sequenced to about 7× redundancy.
Current methods to produce ordered libraries are impractical, because they can cover only short regions (˜5,000 bp) and are labor-intensive.
One
disadvantage to using PCR to amplify the DNA is that only one
genetic element can be amplified in each reaction, unless
multiplex PCR is employed, in which case only as many as 10-50 loci can be simultaneously amplified.
A second disadvantage to PCR is that only a limited number of DNA bases can be amplified from each element (usually <2000 bp).
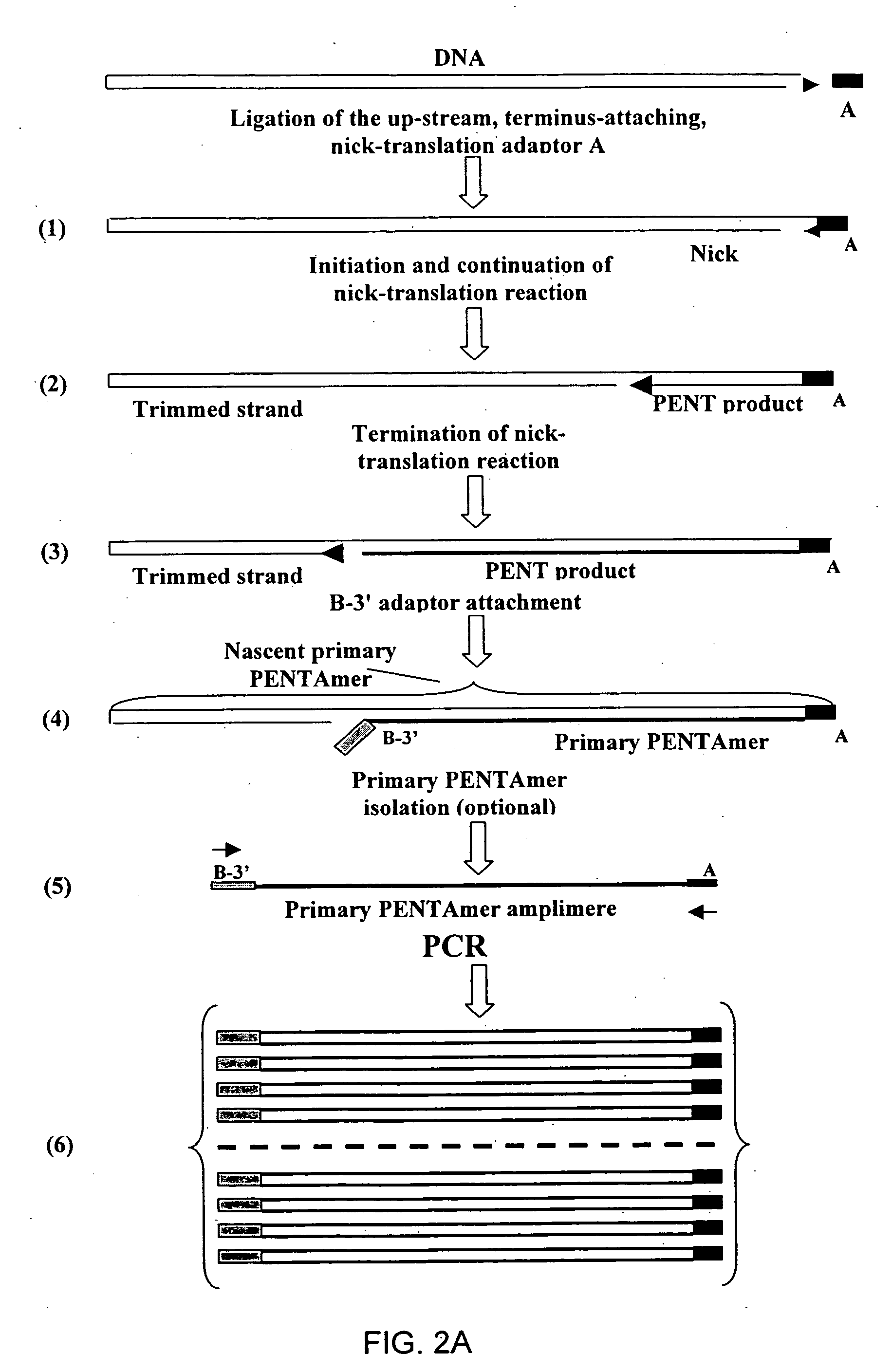
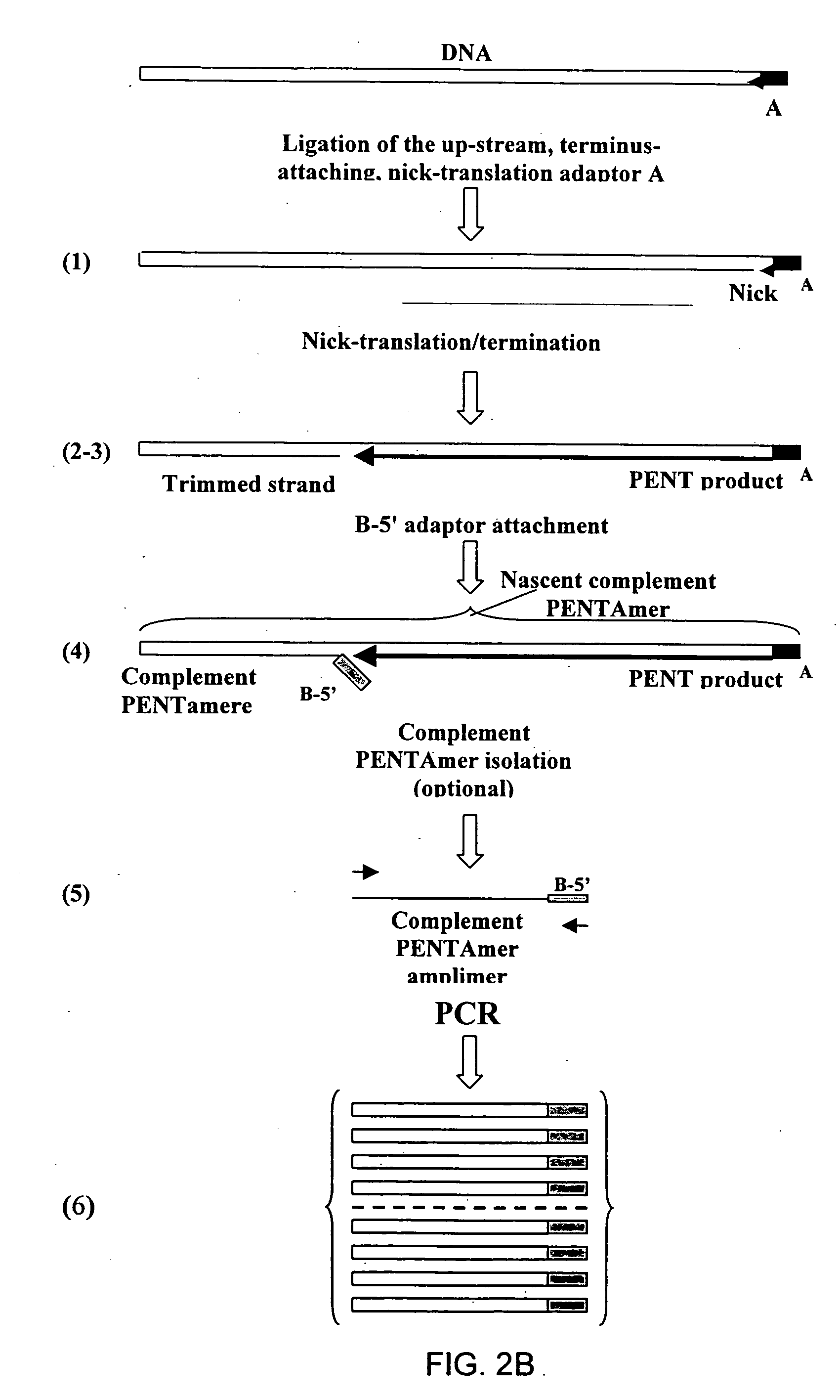
However, this reference fails to teach specific modifications or manipulations prior to the amplification of the nick translation-extended strand to facilitate the amplification.



 Login to View More
Login to View More