However, information present in databases is, to varying degrees, incomplete and inaccurate in relation to the mature expressed protein, due to the multitude of post-translational
processing events that can occur after
protein translation.
Additionally, while the approach is also amenable to the analysis of simple protein mixtures, provided that sufficient
peptide masses are obtained to unambiguously identify each component of the mixture,
peptide mass fingerprinting is generally not suited to the analysis of peptides resulting from
proteolysis of
complex protein mixtures, as the presence of peptides from many different proteins makes it difficult to assign individual peptides to their correct proteins.
Importantly, this method is error tolerant as one, or several, of the regions of the sequence tag (the tag itself or the flanking mass regions) may contain errors due to post-translational modifications yet still result in an unambiguous assignment.
However, as the sequence tag approach requires some user intervention prior to
database analysis, it has not generally been employed for large scale, high
throughput applications.
Hence, the approach is generally considered to be more labor intensive and expensive in terms of both time and sample consumption than the database searching approaches described above.
Unfortunately, several classes of proteins, notably hydrophobic proteins, low abundance proteins, and those with extremes of
isoelectric point (pI) and molecular weight are poorly represented in 2D-
gel based separations (Gygi et al., Proc. Natl. Acad. Sci. U.S.A.
However,
digestion of an unfractionated protein mixture greatly increases the number of components to be analyzed and condenses the
resultant peptide mixture into a narrow mass range, thereby complicating the task of isolating individual components for further analysis, placing greater demands on the performance of the mass
spectrometer.
Furthermore, a problem common to all
peptide sequencing approaches is that MS / MS spectra often yield insufficient product ions, product ions corresponding to cleavages not included in the search algorithms, or lack product ions with sufficient
signal-to-
noise, to allow their identification (Simpson et al.,
Electrophoresis 2000, 21, 1707-1732).
Finally, it is common that many of the peptides resulting from
digestion are not observed, making complete characterization of the protein difficult to achieve.
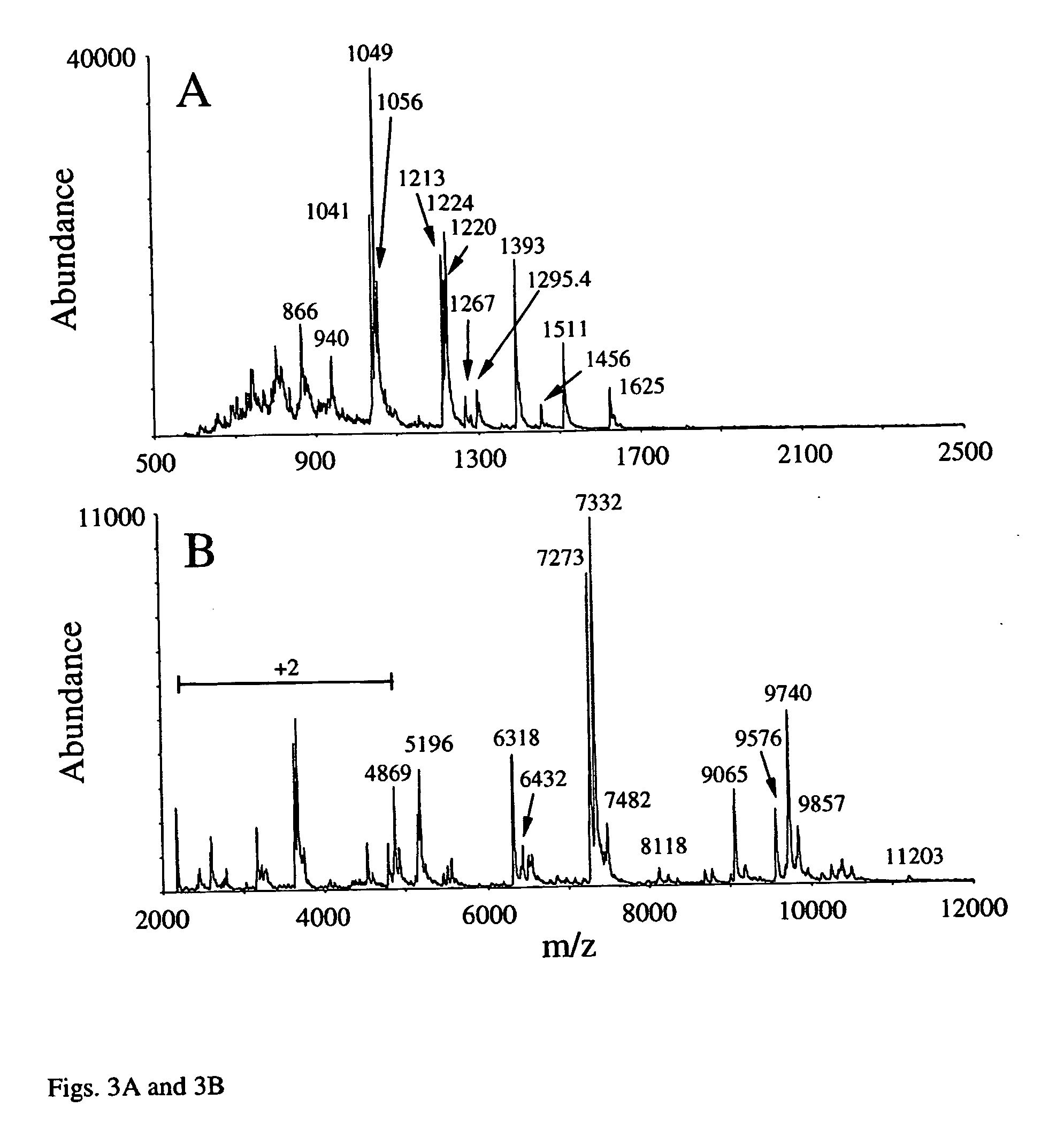
While protein mass is fundamentally informative, it alone is not particularly useful in unambiguously identifying a protein due to the potential for post-translational
processing to cause differences between the predicted and experimentally observed masses.
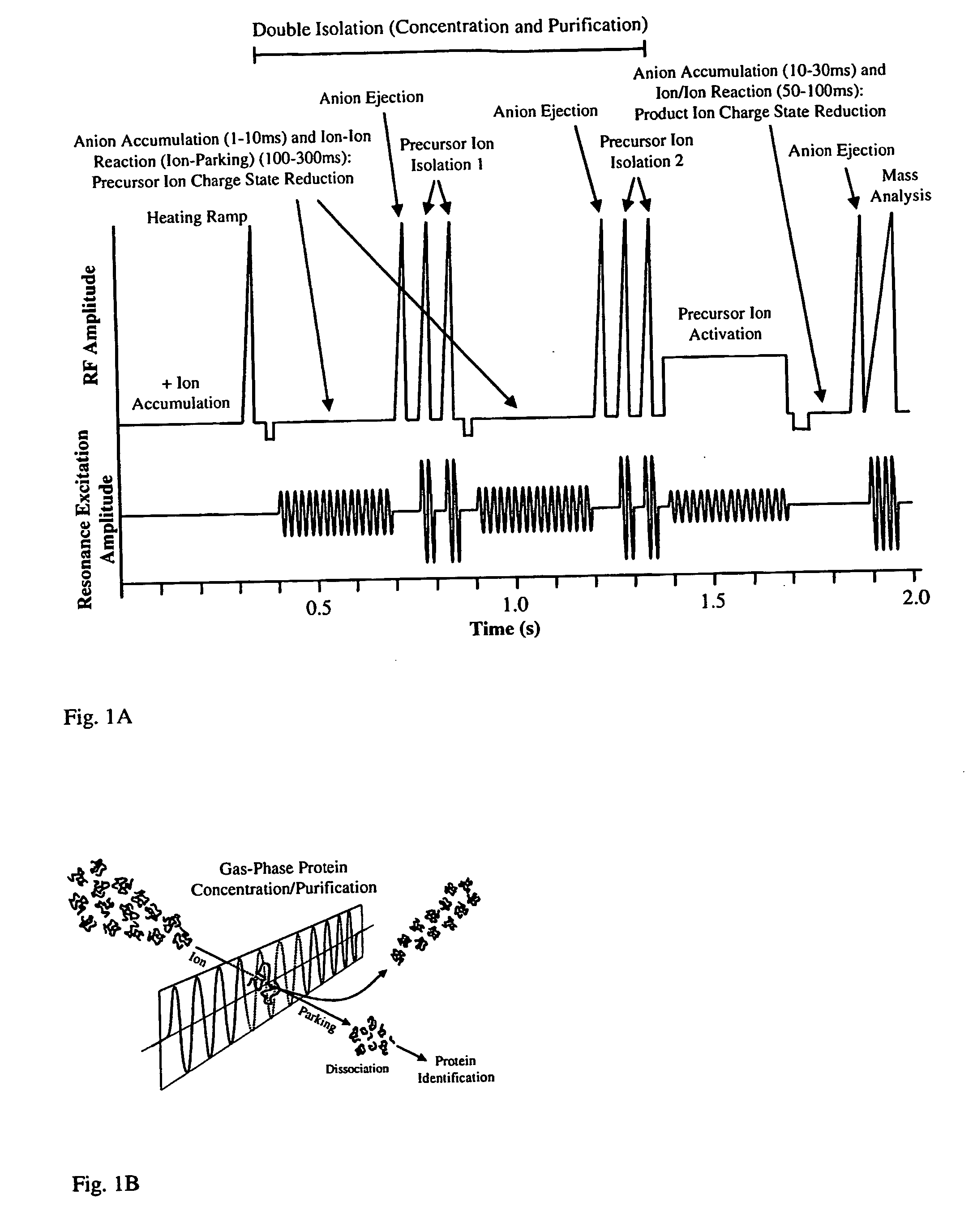
A major issue associated with implementation of the top-down approach on most types of tandem mass spectrometers is that the spectra derived from the dissociation of multiply-charged proteins ions typically contain product ions with charge states
ranging from +1 up to the charge of the precursor ion, thereby creating possible ambiguities in assigning product ion mass and charge.
A limitation of the “double isolation” approach is that the ions from a given protein charge state are distributed over several charge states during the ion / ion reaction, thereby diluting the protein ion
signal and decreasing the sensitivity for subsequent isolation and dissociation.
Although techniques that increase “charge state purification” such as double isolation of the precursor ion and ion parking have made the “top down” approach to
mass spectrometry-driven
protein sequencing more reliable, difficulties in matching product ion spectra with
protein database information still frequently arise, especially when the information in databases is incomplete and inaccurate in relation to the mature expressed protein.
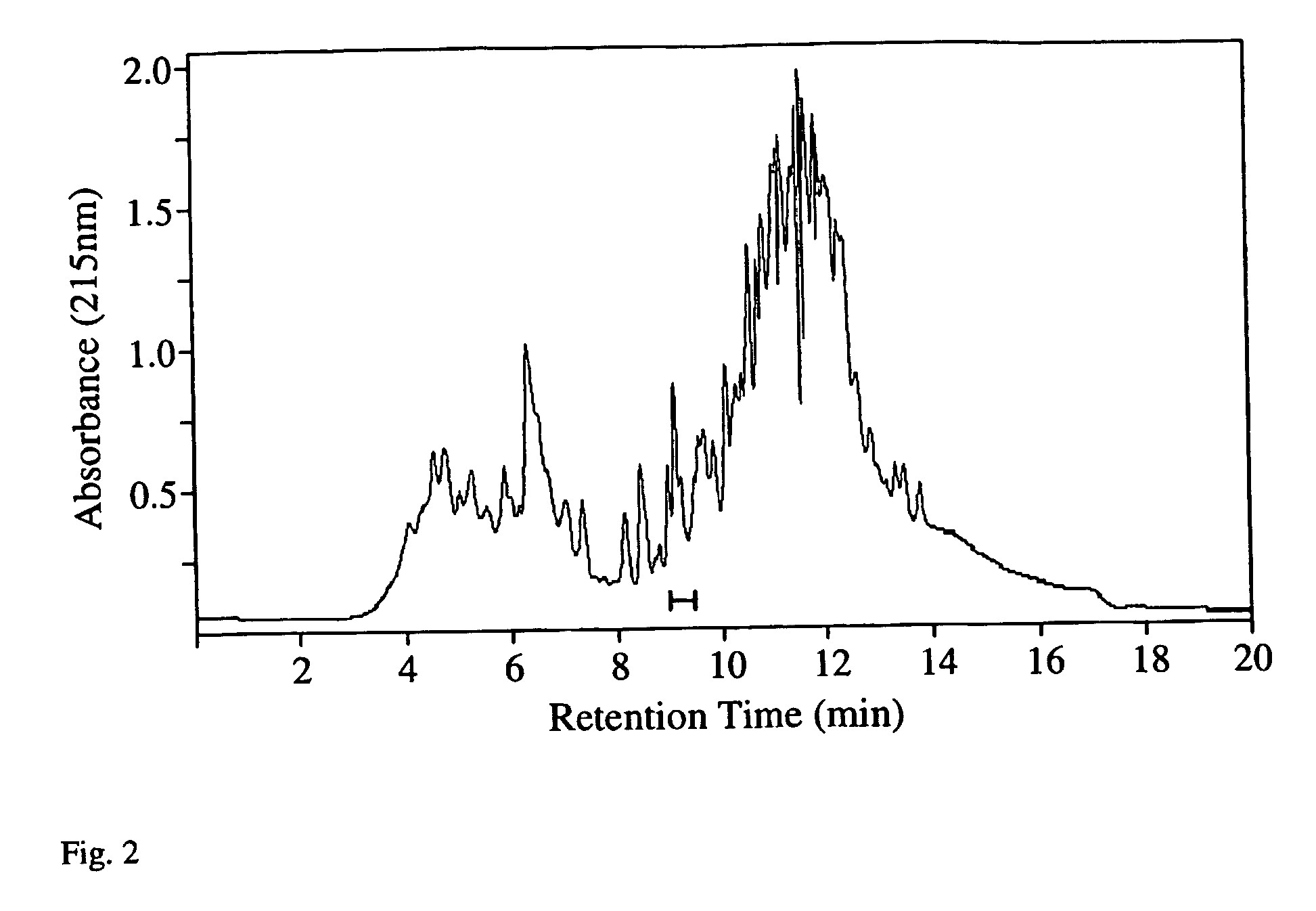
The identification and characterization of proteins present in complex mixtures remains an important
analytical problem.



 Login to View More
Login to View More