

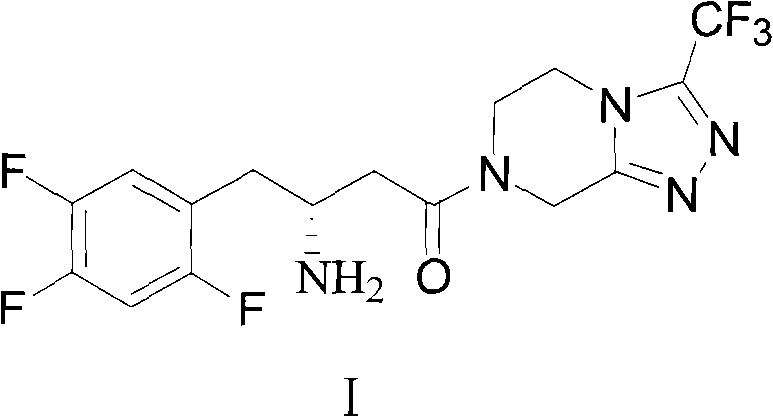
Preparation method of sitagliptin intermediate, sitagliptin or salts thereof
A technology for sitagliptin and intermediates, which is applied in the preparation of organic compounds, the preparation of carboxylic acid amides, chemical instruments and methods, etc., can solve the problems of unstable product quality, high cost and high cost, and avoid the problem of low synthesis efficiency. Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
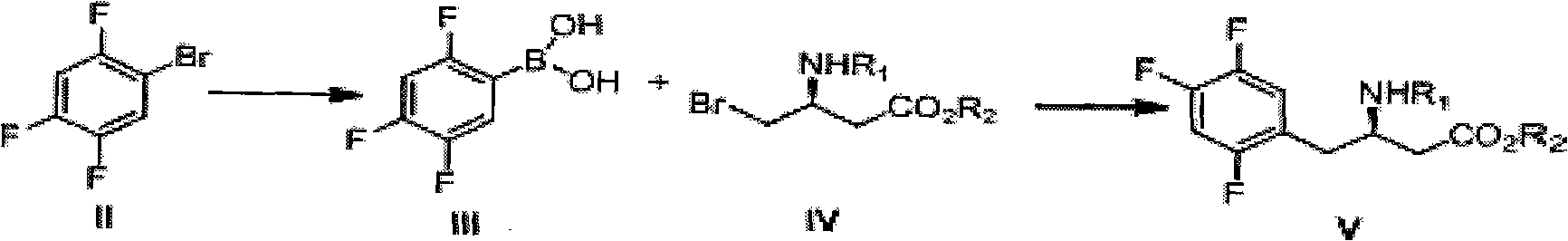
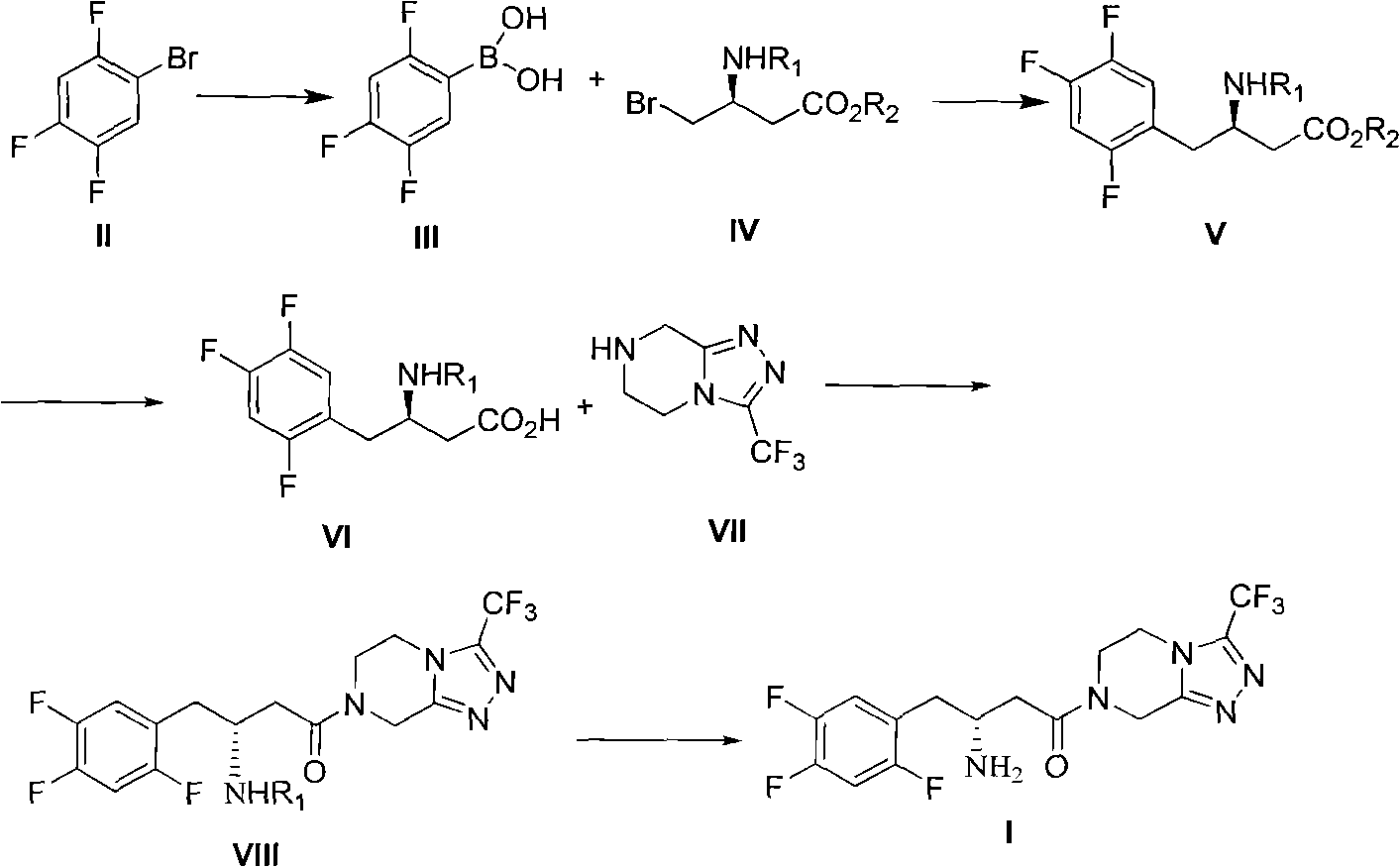
[0045] Preparation of (3R)-3 tert-Butoxycarbonylamino-4-bromo-butyric acid methyl ester (IV)
[0046] In a 250 ml round bottom flask, add (3R)-3 tert-butoxycarbonylamino-4-hydroxy-butyric acid methyl ester (IX) (23.3 g, 0.1 mol) and 150 ml of dichloromethane, and then add triphenyl Phosphine (26.2g, 0.1mol), bromine (17.6g, 0.11mol) was added dropwise at room temperature, and the drop was finished within ten minutes. After the addition, continue to stir for 3 hours. The reaction was quenched with 10% sodium bisulfite aqueous solution, liquid separation, the organic phase was washed with saturated brine, dried, filtered, and concentrated to obtain a crude product, which was then distilled under reduced pressure to obtain the product (26g, 0.088mol) ), the yield is 88%. 1 H NMR(300MHz, CDCl 3 ) 1.48 (9H, s), 2.67 (1H, d, J = 6.4 and 16.5), 2.80 (1H, dd, J = 5.5 and 16.5), 3.22 (2H, br), 3.78 (3H, s), 5.00 ( 1H, m), and 5.40 (1H, br); ESI-MS m / z 296.2 (M+H) + .
Embodiment 2
[0048] Preparation of 2,4,5-trifluorophenylboronic acid (III)
[0049] In a 250 ml three-necked flask, add a smooth surface of magnesium bar (3.96g, 0.165mol) and a small particle of iodine, 2,4,5-trifluorobromobenzene (II) (31.5g, 0.15mol) with 60 ml After diluting with water and ether, adding a small amount of dropwise to initiate the reaction, add the remaining ether solution dropwise to the reaction flask, keeping the dropping rate at the same rate as the reflux rate, and heating to reflux after the addition. At 4 hours, the magnesium bar basically reacted. In another 500ml three-necked flask, add trimethyl borate (15.6g, 0.15mol) and 150ml anhydrous tetrahydrofuran, under the protection of nitrogen, cool to -78℃, and add the prepared grating reagent dropwise to the rapidly stirring Trimethyl borate solution. After holding at -78°C for 1 hour, it will naturally rise to room temperature, add 150 ml of water and 150 ml of 20% citric acid, and stir for 1 hour. The tetrahydrof...
Embodiment 3
[0051] Preparation of (3R)-3 tert-butoxycarbonylamino-4-(2,4,5-trifluorophenyl)-butyric acid methyl ester (V)
[0052] In a 250 ml three-necked flask, add (3R)-3 tert-butoxycarbonylamino-4-bromo-butyric acid methyl ester (IV) (14.75g, 0.05mol), 2,4,5-trifluorophenylboronic acid ( III) (10.56g, 0.06mol), potassium tert-butoxide (16.8g, 0.15mol), Pd(OAc) 2 (0.224g, 0.001mol), 100ml of 1,4-dioxane. Under nitrogen protection, stir at 50°C for 10 hours. Cool to room temperature, evaporate the solvent, add 200 ml of ethyl acetate, 200 ml of water, separate the liquids, wash the organic phase with 10% dilute hydrochloric acid, then wash with saturated brine, dry, filter, concentrate to obtain a crude product, and recrystallize from toluene White solid (13g, 0.0375mol), yield 75%. Compound physical and chemical data and literature [1] Same as described in.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More