

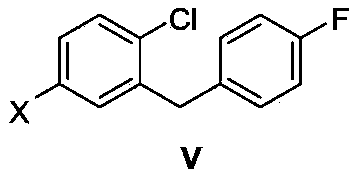
4-substituted-1-chloro-2-(4-fluorobenzyl)benzene, its preparation method and application as intermediate in preparation of anti-type II diabetes drugs
A kind of fluorobenzyl, C1-C3 technology, applied in 4-substituted-1-chloro-2-(4-fluorobenzyl)benzene and its preparation and application field in the preparation of anti-type II diabetes drugs as an intermediate , which can solve the problems of limited synthesis routes, and achieve the effects of good yield, simple and safe operation, and good economic effect.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

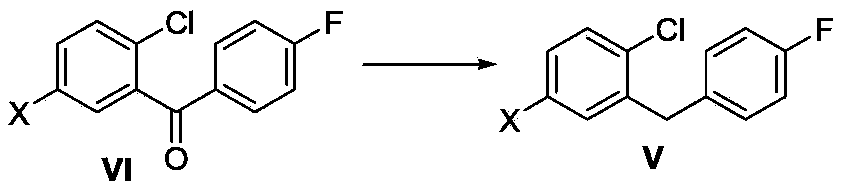
Examples
example 14
[0029] The synthesis of example 14-bromo-1-chloro-2-(4-fluorobenzyl)benzene (Va)
[0030] Add (5-bromo-2-chlorophenyl)(4-fluorophenyl)methanone (VIa, 31.3g, 100mmol) and 190mL of dichloromethane into a 500mL three-necked flask, stir and dissolve at room temperature, then cool down to 0-5°C , slowly drop triethylsilane (44.2g, 380mmol), then slowly add boron trifluoride diethyl ether (51.8g, 365mmol) dropwise at a controlled temperature between 0 and 5°C, and return to 20-30°C for 4-6 Hours, HPLC traced to raw material remainder<3%. Add 150 mL of saturated aqueous sodium bicarbonate solution to the reaction flask, stir for 30 minutes, and separate the liquids. The organic phase was concentrated until no distillate flowed out, 230 g of deionized water was added, stirred until solid precipitated, and filtered. Dissolve the solid in 110mL of acetonitrile, add dropwise 110mL of 2% sodium hydroxide aqueous solution, stir, filter the system, wash the filter cake with a little water...
example 2
[0032]Synthesis of Example 21-chloro-2-(4-fluorobenzyl)-4-iodobenzene (Vb)
[0033] Add (2-chloro-5-iodophenyl)(4-fluorophenyl)methanone (VIb, 36.1g, 100mmol) and 180mL acetonitrile into a 500mL three-necked flask, stir and dissolve at room temperature, then cool down to 0-5°C, slowly Add triethylsilane (44.2g, 380mmol) dropwise, then slowly add boron trifluoride diethyl ether (51.8g, 365mmol) dropwise at a controlled temperature of 0-5°C, return to 20-30°C and react for 4-6 hours, HPLC Track until the remaining raw material is less than 3%. Add 150 mL of saturated aqueous sodium bicarbonate solution to the reaction flask, stir for 30 minutes, and separate the layers. Concentrate the organic phase to 95-110 mL, add 230 g of deionized water, stir until solid precipitates, and filter. The solid was rinsed with acetonitrile / water=1 / 3 system (100mL), and dried to obtain an off-white solid, namely the compound 1-chloro-2-(4-fluorobenzyl)-4-iodobenzene (Vb) (27.4 g, yield 79%). ...
example 3
[0035] Example 3 (2S, 3R, 4S, 5R, 6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-2-(4-chloro-3-(4-fluorobenzyl Synthesis of (yl)phenyl)-2-methoxytetrahydro-2H-pyran (IVa)
[0036] Method 1): Add 4-bromo-1-chloro-2-(4-fluorobenzyl)benzene (Va, 3g, 10mmol) and 15mL THF into a three-necked flask, cool down to -78°C under nitrogen protection, and slowly add positive Butyl lithium in n-heptane (10 mL, 10 mmol) was added dropwise, and stirred at -78°C for 1 hour. Add (3R,4S,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)tetrahydro-2H-pyran-2-one dropwise at -78°C THF solution (VIIa, 5.4g, 10mmol, dissolved in 6mL THF), the system was reacted at -78°C for 1 hour. After that, 20 mL of methanol solution containing 1.5 mL of methanesulfonic acid was added, and the system was warmed to room temperature and stirred overnight. Add saturated aqueous sodium bicarbonate solution to the system to quench the reaction, separate the layers, extract the aqueous phase twice with 50 mL of ethyl a...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More