

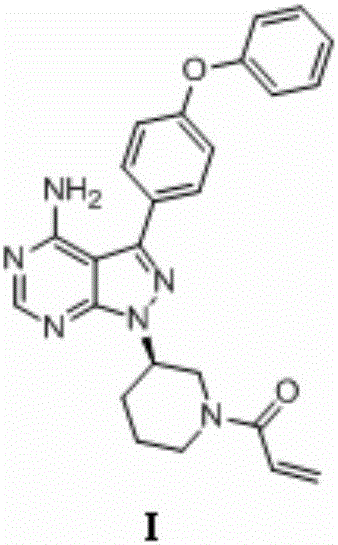
Preparation method of ibrutinib
A technology of ibrutinib and compound, applied in the field of preparation of ibrutinib, can solve the problems of unfavorable industrial scale, inconvenient operation, high price, etc., achieve stable and controllable synthesis process, easy storage, shorten separation and purification effect of steps
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1
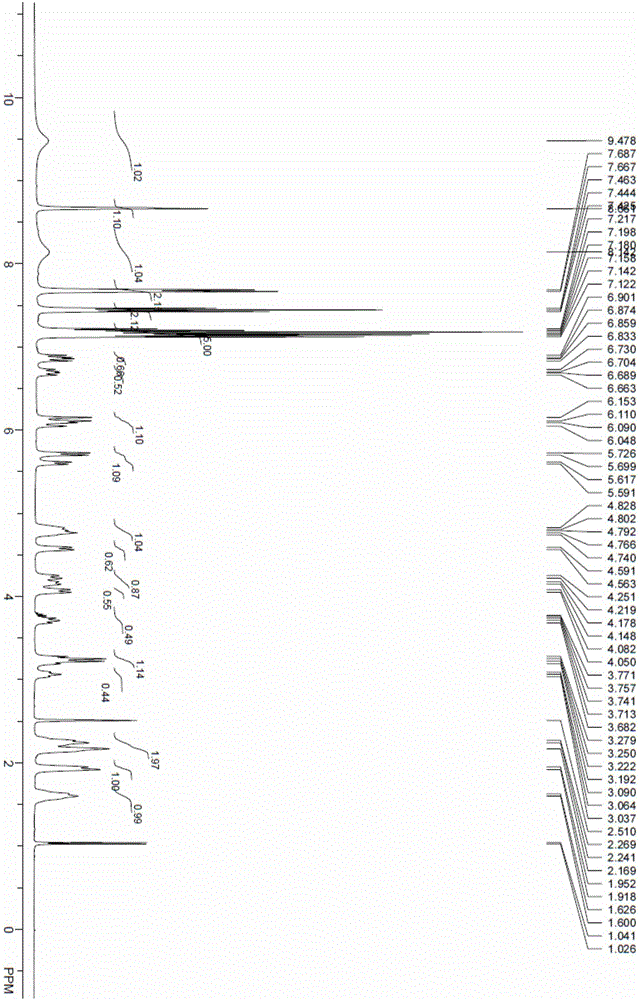
[0061] Synthesis of (R)-3-(4-phenoxyphenyl)-1-(piperidin-3-yl)-1H-pyrazolo(3,4-d)pyrimidin-4-amine (5)
[0062] Add 20 g (66 mmol) of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine into a 1L three-necked flask; S-1-tert-butoxycarbonyl-3- Hydroxypiperidine 13.3g (66mmol); triphenylphosphine 34.6g (132mmol), add dry tetrahydrofuran 300mL, cool to 5-10°C. Under nitrogen protection, 26.7 g (132 mmol) of diisopropyl azodicarboxylate was slowly added dropwise into the bottle, and the reaction was carried out at 30° C. for 10 hours after the drop was completed. The reaction solution was re-cooled to 0-10°C, 13.4g (132mmol) of 36% hydrochloric acid was added dropwise, and the temperature was raised to 40°C for 1 hour after the dropwise completion. Add water, stir, extract 3 times with dichloromethane, collect the water phase, adjust the water phase to be alkaline (pH>10) with sodium hydroxide solution, precipitate the solid, filter, collect the solid, and bake at 50°C for 8...
Embodiment 2
[0064] Synthesis of (R)-3-(4-phenoxyphenyl)-1-(piperidin-3-yl)-1H-pyrazolo(3,4-d)pyrimidin-4-amine (5)
[0065] Add 20 g (66 mmol) of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine into a 1L three-necked flask; S-1-tert-butoxycarbonyl-3- Hydroxypiperidine 26.6g (132mmol); triphenylphosphine 51.9g (198mmol), add dry tetrahydrofuran 400mL, cool to 5-10°C. Under nitrogen protection, 40 g (198 mmol) of diisopropyl azodicarboxylate was slowly added dropwise to the bottle, and the reaction was carried out at 30° C. for 12 hours after the drop was completed. The reaction solution was re-cooled to 0-10°C, 20 g (198 mmol) of 36% hydrochloric acid was added dropwise, and the temperature was raised to 40°C for 1 hour after the dropwise completion. Add water, stir, extract 3 times with dichloromethane, collect the water phase, adjust the water phase to be alkaline (pH>10) with sodium hydroxide solution, precipitate the solid, filter, collect the solid, and bake at 50°C for 12 h...
Embodiment 3
[0067] Synthesis of (R)-3-(4-phenoxyphenyl)-1-(piperidin-3-yl)-1H-pyrazolo(3,4-d)pyrimidin-4-amine (5)
[0068] Add 20 g (66 mmol) of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine into a 1L three-necked flask; S-1-tert-butoxycarbonyl-3- Hydroxypiperidine 26.6g (132mmol); triphenylphosphine 51.9g (198mmol), add dry tetrahydrofuran 400mL, cool to 5-10°C. Under nitrogen protection, 40 g (198 mmol) of diisopropyl azodicarboxylate was slowly added dropwise to the bottle, and the reaction was carried out at 30° C. for 12 hours after the drop was completed. The reaction solution was re-cooled to 0-10°C, 22.5 g (198 mmol) of trifluoroacetic acid was added dropwise, and the temperature was raised to 40°C for 1 hour after the dropwise completion. TLC monitored the end of the reaction. Add water, stir, extract 3 times with dichloromethane, collect the water phase, adjust the water phase to be alkaline (pH>10) with sodium hydroxide solution, precipitate the solid, filter, col...
PUM
| Property | Measurement | Unit |
|---|---|---|
| optical purity | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More