

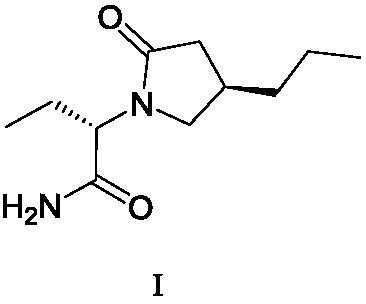
A kind of preparation method of furanone compound
A compound and furanone technology, applied in the field of preparation of furanone compounds, can solve the problems of low product purity, high production cost, unsuitable for industrial production and the like
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0066]
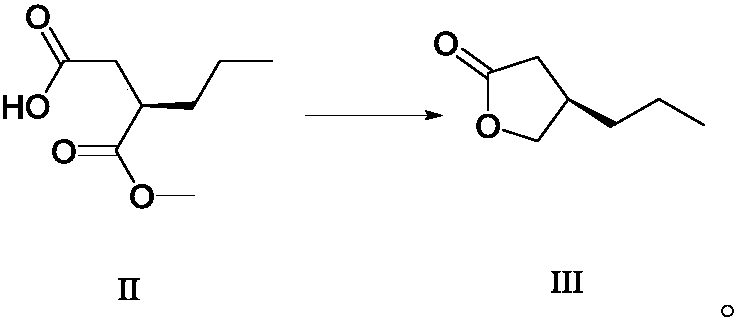
[0067] Dissolve (R)-3-methoxycarbonylhexanoic acid (174.1g, 1mol, ee value 99.2%) in 500mL of methanol, cool down to 0°C, add 500mL of water, cool to 0°C, add powdered chlorine Calcium chloride (115.8 g, 1.1 mol) and sodium borohydride in ethanol (2M, 800 mL). The reaction solution was stirred overnight (about 12 hours) at (20°C-30°C) and then quenched by adding hydrochloric acid (6M, 1000mL), concentrated under reduced pressure, diluted with 500mL of water, extracted with dichloromethane (3×150mL), and combined The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain 108.9 g of furanone compound III with a yield of 85.0% and a purity of 97.2% (GC).
[0068] R-3-methoxycarbonylhexanoic acid can be prepared according to the method described in Angewandte Chemie International Edition, 1998, 37(13-14), 1931-1933, and the ee value is greater than 99.0%.
Embodiment 2
[0070]
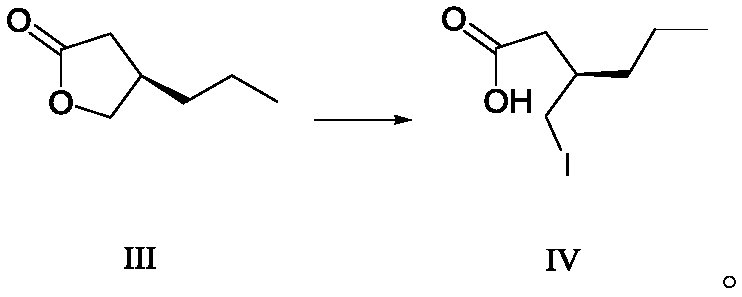
[0071] Under nitrogen protection, furanone compound III (128.1 g, 1 mol) was dissolved in 1 L of dichloromethane, cooled to 0 ° C, trimethyl iodosilane (150 mL) was added, and the reaction solution was stirred at 20 to 30 ° C for 2 Hour. Then add hydrochloric acid solution (1M, 800mL) and sodium thiosulfate aqueous solution (mass percentage is 10%, described mass percent refers to the percentage of the quality of sodium thiosulfate and sodium thiosulfate aqueous solution gross mass, 400mL) successively. , the aqueous phase was extracted with 1L dichloromethane, the organic phase was washed twice with saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain Buvaracetam intermediate IV (254.6g), yield 99.5%, purity : 95.6% (GC).
Embodiment 3
[0073]
[0074] Under the protection of nitrogen, dissolve Buvaracetam intermediate IV (1280.4g, 4mol) in 1500mL toluene, slowly add thionyl chloride (951.8g, 8mol), and stir the reaction solution at room temperature (20°C~30°C) After 24 hours, the solvent was concentrated under reduced pressure. The residue was rectified under vacuum pump (0.32 mmHg, 90-95° C.) to obtain 1310 g of light yellow transparent liquid Compound V.
[0075] 1310 g of the obtained compound V was dissolved in 2.5 L of dichloromethane solution. Subsequently, the above solution was added to a mixture containing L-2-aminobutyramide hydrochloride (428.9g, 4.2mol), 4A molecular sieves (500g), potassium hydroxide (500g), anhydrous sodium sulfate (500g), tetrabutyl bromide In dichloromethane solution (12.5L) of ammonium (49g, 0.14mol), the reaction solution was stirred at 20-30°C for 18 hours, then filtered with diatomaceous earth, and the filtrate was concentrated to dryness under reduced pressure to obt...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More