

Preparation method of Roxadustat
A technology of roxadustat and compounds, which is applied in the field of preparation of roxadustat, can solve the problems of unsuitability for industrial production, difficulty in obtaining starting materials, and high reaction temperature, and achieve no safety hazard, short reaction time, and simple operation Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0074] Example 1: Methyl 1-bromo-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate
[0075]
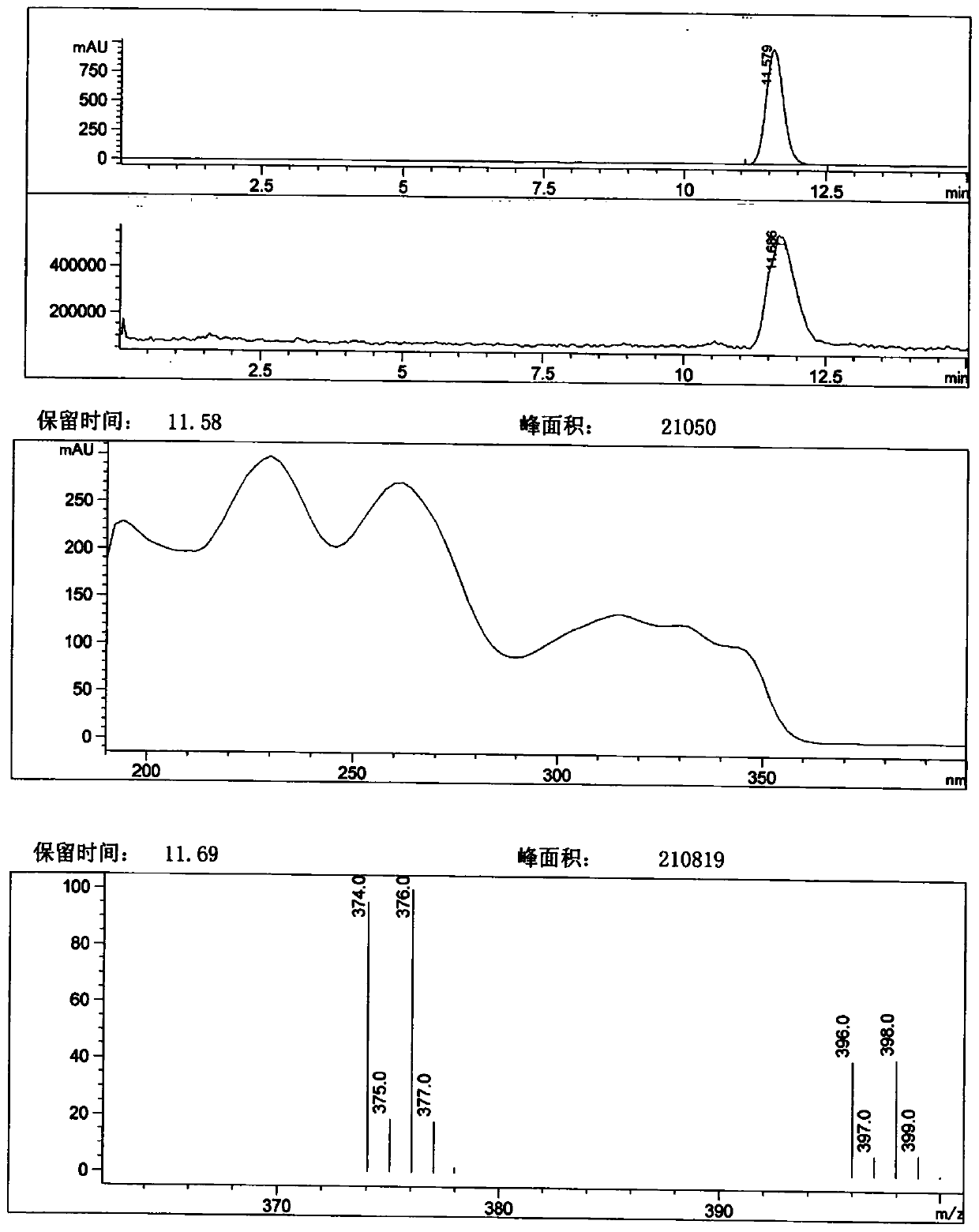
[0076] Methyl 4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (50.00g, 169.45mmol) was added to 500ml of tetrahydrofuran, then NBS (31.67g, 177.93mmol) and AIBN (1.39g, 8.47mmol) ). After the addition, react at 25°C for 1h. TLC analysis (petroleum ether-ethyl acetate=3:2) showed that the reaction was complete. Post-treatment: Concentrate under reduced pressure to remove most of the solvent, then add 500ml of methanol and heat to reflux for 0.5h, cool down, filter, wash the filter cake with about 50ml of methanol, and dry to obtain 60.60g of off-white solid, yield 95.64%, LC- MS spectrum see figure 1 , The spectrum shows no obvious impurity peaks.
[0077] 1 H-NMR(600MHz, DMSO-d 6 ): δ3.00 (3H, s), 7.27 (2H, dd, J=7.2, 1.2Hz), 7.34 (1H, t, J=7.2Hz), 7.41 (1H, d, J=2.4Hz), 7.54 (2H, td, J=7.2, 1.2 Hz), 7.69 (1H, dd, J=9.0, 2.4 Hz), 8.37 (1H, d, J=9.0 Hz), 11.54 (1H, s).
Embodiment 2
[0078] Example 2: 1-Bromo-4-hydroxy-7-phenoxyisoquinoline-3-carboxylic acid
[0079]
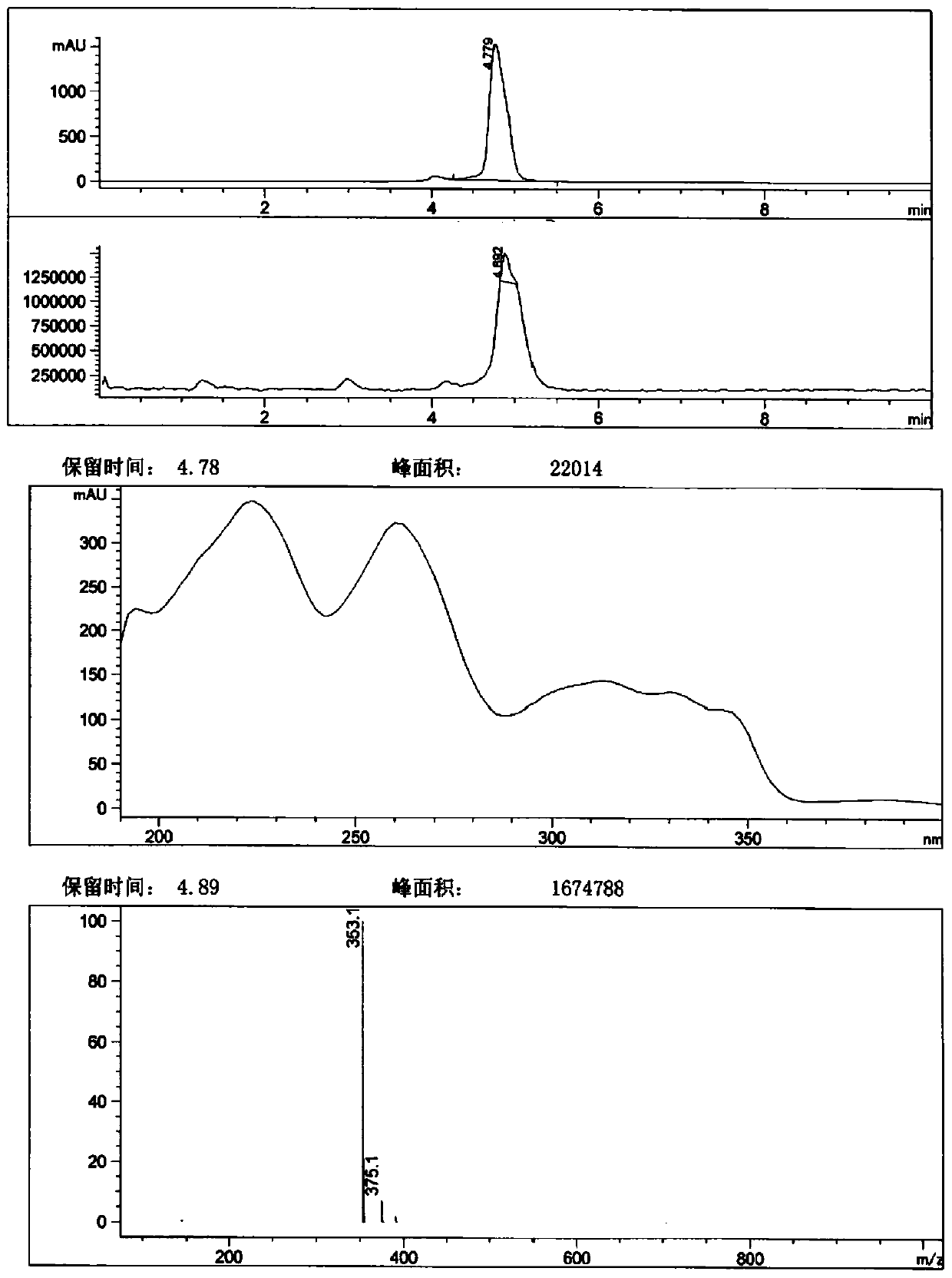
[0080] Methyl 1-bromo-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (71.00g, 189.74mmol) was added to 300ml of ethanol, then sodium hydroxide (22.71g, 569.22mmol) and 300ml were added A mixture of water. After the addition, the temperature is raised to reflux for about 1 hour. TLC analysis (petroleum ether-ethyl acetate=5:1) showed that the reaction was complete. Post-treatment: cool down, concentrate under reduced pressure to remove part of the solvent, then add 500ml of water, 2M hydrochloric acid to adjust the pH to 2-3, stir and crystallize for 0.5h, filter, and dry to obtain 67.70g of light red solid, related substances 99.31%, yield 99.06%.
[0081] 1 H-NMR(600MHz, DMSO-d 6 ): δ 7.27 (2H, dd, J=7.2, 1.2 Hz), 7.34 (1H, t, J=7.2 Hz), 7.42 (1H, d, J=2.4 Hz), 7.54 (2H, td, J= 7.2, 1.2 Hz), 7.68 (1H, dd, J=9.0, 2.4 Hz), 8.36 (1H, d, J=9.0 Hz), 13.08 (about 2H, brs).
Embodiment 3
[0082] Example 3: (1-Bromo-4-hydroxy-7-phenoxyisoquinoline-3-carbonyl)glycine methyl ester
[0083]
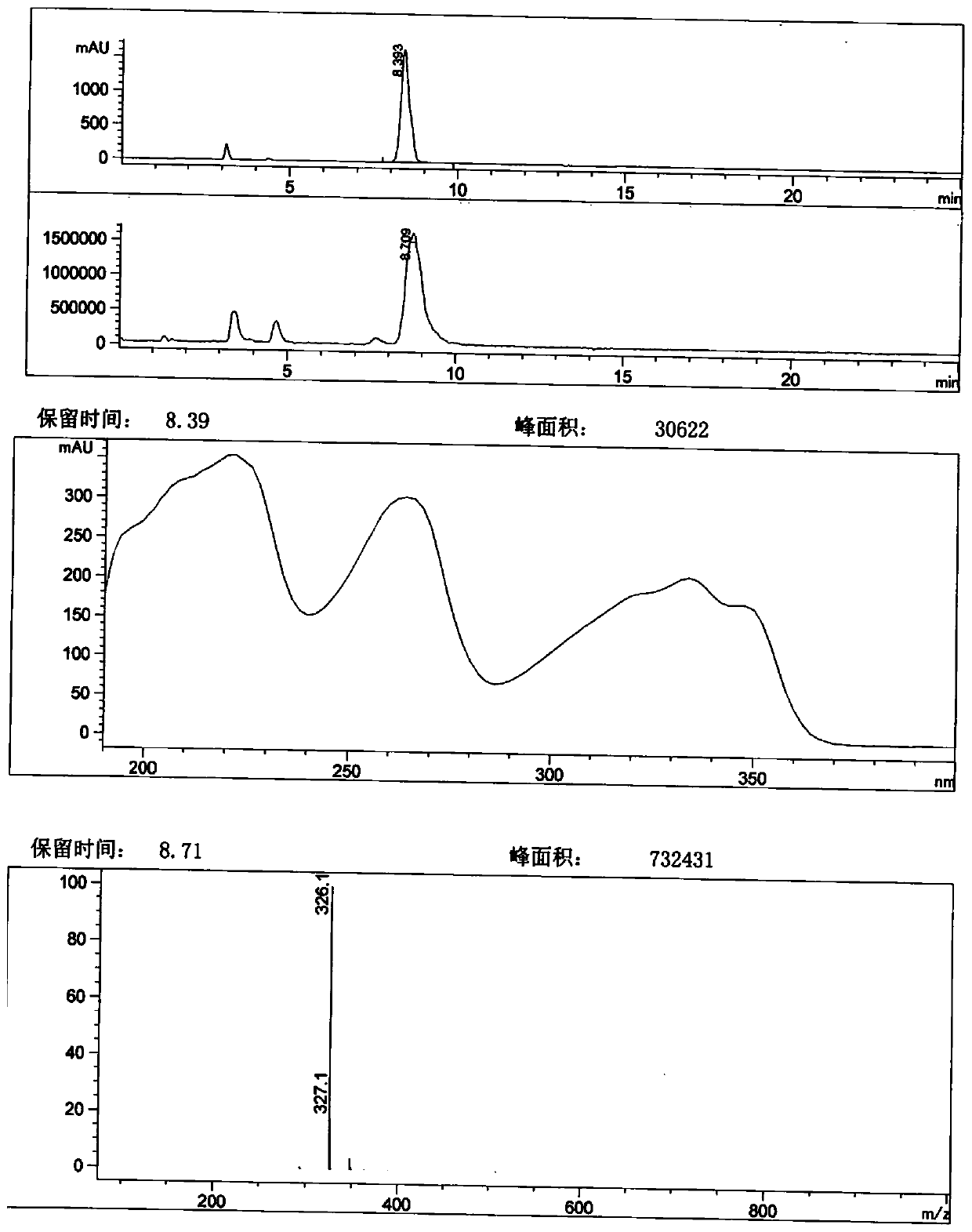
[0084] Add 1-bromo-4-hydroxy-7-phenoxyisoquinoline-3-carboxylic acid (20.00g, 55.53mmol) to 250ml of dichloromethane, and then add glycine methyl ester hydrochloride (10.46g, 83.30mmol) ) And PyBOP (43.35g, 83.30mmol), and finally triethylamine (22.48g, 222.12mmol) was added. After the addition, the temperature is controlled at 25°C to react for about 2h. TLC analysis (DCM-MeOH (10:1)+3dHAc) showed that the reaction was almost complete. Post-treatment: add 120ml of water to the reaction system, separate the liquids, extract twice with dichloromethane (200ml×2), combine the organic phases, dry with anhydrous magnesium sulfate, decolorize with activated carbon, filter, and concentrate under reduced pressure to obtain 75.02g of a white solid.
[0085] Refining: add 400ml of isopropanol to the above crude product, heat and reflux for 0.5h, cool down, filter, wash the filter cake wit...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More