

Process for preparing β-artemether
A technology of artemether and methanesulfonic acid, applied in the field of synthesis of β-artemether, can solve the problems such as that the yield of β-artemether cannot be further improved, and achieves easy control of process operation, high product quality and low cost Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

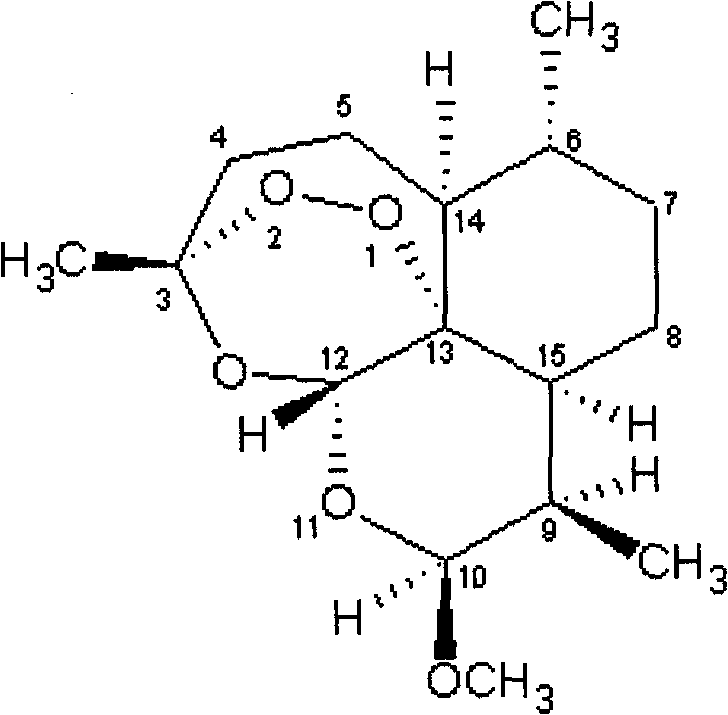
Examples
preparation example Construction
[0021] The method for preparing β-artemether of the present invention only uses trimethyl orthoformate as the etherifying agent.
[0022] The method for preparing β-artemether of the present invention comprises the following steps: taking dihydroartemisinin as a raw material, and performing etherification reaction with trimethyl orthoformate in an organic solvent to obtain β-artemether.
[0023] The organic solvent is selected from ester solvents or alkane solvents.
[0024] The ester solvent is selected from methyl formate, ethyl formate, methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobutyl acetate, dimethyl carbonate, diethyl carbonate, Propyl carbonate or isopropyl carbonate. Methyl acetate or ethyl acetate is preferred.
[0025] The alkane solvent is selected from n-hexane, n-heptane or petroleum ether.
[0026] The catalyst is selected from boron trifluoride ether, trifluoroacetic acid, p-toluenesulfonic acid or methanesulfonic acid...
Embodiment 1
[0032] Under nitrogen protection, dihydroartemisinin (40.0 g, 0.14 mol) was added to 150 ml of ethyl acetate and trimethyl orthoformate (10.5 ml, 0.096 mol); cooled in an ice bath, the temperature of the system was lowered to 0 At °C, a solution of boron trifluoride ether (2.0 ml, 0.016 mol) in ethyl acetate (50 ml) was added dropwise. After the dropwise addition, the reaction was continued at 0°C for 2 hours. The ice bath was removed, the temperature was naturally raised to room temperature, and the reaction was continued for 1 hour. The reaction was followed by TLC. After confirming that the reaction was complete, the temperature was cooled to 5° C., and a saturated sodium bicarbonate solution was added dropwise to adjust the pH to 7-8. The layers were separated, the organic layer was washed once with saturated brine, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to obtain about 43 g of semi-solid.
[0033] 43 g of the obtained crude product ...
Embodiment 2
[0035] Dihydroartemisinin (40 g, 0.14 mol) was added to 150 ml of methyl acetate and trimethyl orthoformate (12.0 ml, 0.11 mol); cooled in an ice bath, the temperature of the system was lowered to 5 °C, and methanesulfonic acid was added dropwise Acid (1.0 mL, 7.9 mmol) in methyl acetate (50 mL). After the dropwise addition was completed, the reaction was continued at 5°C for 1 hour. The ice bath was removed, the temperature was naturally raised to room temperature, and the reaction was continued for 1 hour.
[0036] The post-processing method was the same as that in Example 1, and 38.2 g of β-artemether was obtained, with a molar yield of 91.5%, a purity of 99.5% detected by HPLC, and a single impurity of less than 0.1%. NMR data as above.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More