

Method for preparing halofuginone hydrobromide
A technology of permanenone hydrobromide and piperidone, which is applied in the field of preparation of permanenone hydrobromide, can solve the problems of three waste pollution process routes, complex reaction conditions, cumbersome synthesis steps, etc., and achieve cost reduction, good product purity, The effect of reducing the pollution of three wastes
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


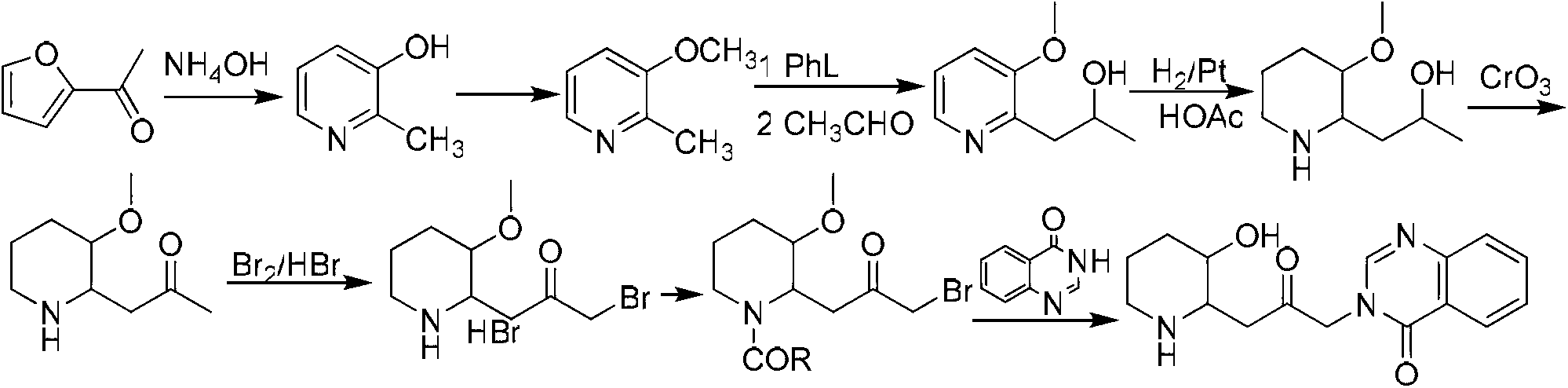
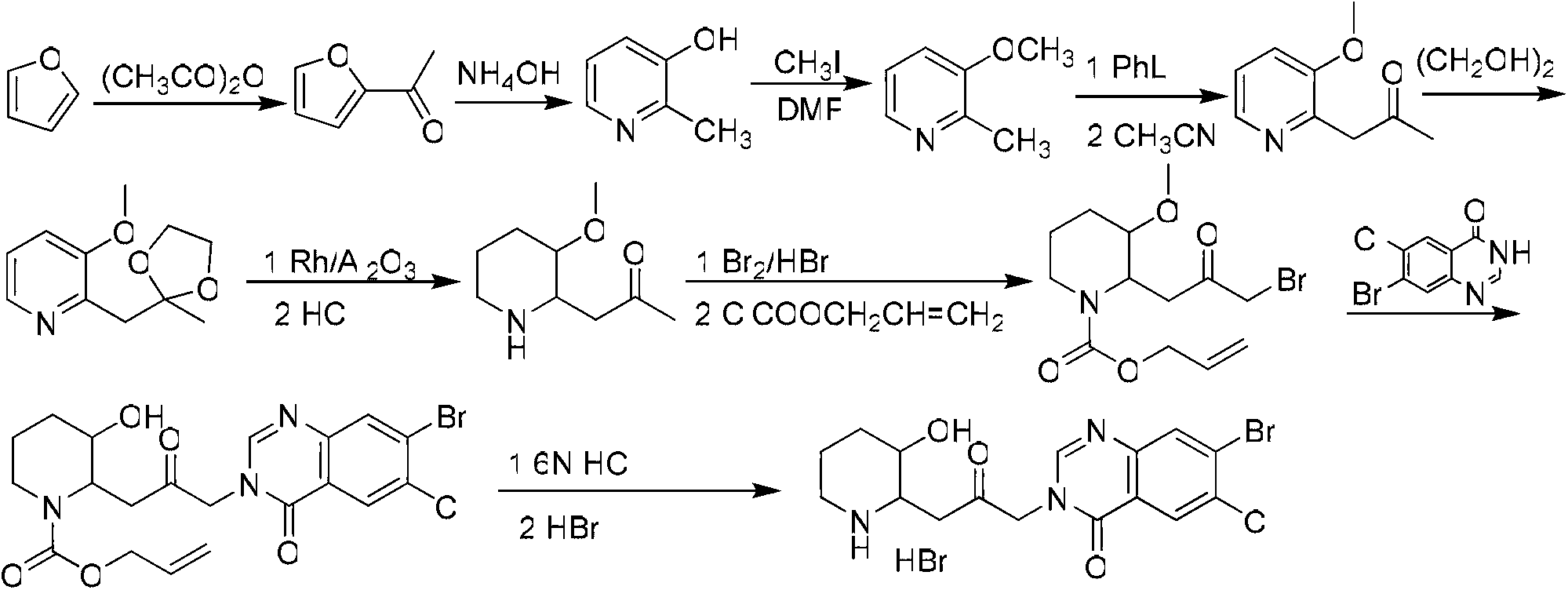
Examples
Embodiment 1
[0064] Preparation of Intermediate VIII
[0065] Add anhydrous potassium carbonate (13.71g, 0.99mol), anhydrous sodium iodide (14.90g, 0.99mol), acetonitrile 200mL, N-benzyl-3-piperidone (17.12g , 0.90mol), 2,3-dichloropropene (14.83g, 1.35mol), a reflux condenser was installed, heated and stirred to reflux for 13 hours. Cool, add 20 mL of water, recover acetonitrile by distillation, add 100 mL of water, extract with ethyl acetate (100 mL×3), combine the ethyl acetate extracts, wash once with 50 mL of saturated saline, dry over anhydrous sodium sulfate, filter, and evaporate ethyl acetate The ester obtained 19.7 g of yellow viscous liquid, ie intermediate VIII, with a yield of 83%.
[0066] R f =0.6(petroleum ether:EtOAc=3:1); IR(KBr)v max : 2951, 1716, 1635, 1454, 1136, 885cm -1 ; 1 H NMR (400MHz, CDCl 3 ): δ1.97-2.04(m, 2H), 2.38-2.43(m, 1H), 2.45-2.57(m, 1H), 2.61-2.67(m, 1H), 2.84-2.86(m, 2H), 2.90 -2.95(m, 1H), 3.52(t, 1H, J=6.4Hz), 3.62(d, 1H, J=13.4Hz), 3.80(d, 1...
Embodiment 2
[0088] The preparation of embodiment 2 intermediate VIII
[0089] Add anhydrous sodium carbonate (10.49g, 0.099mol), acetonitrile 200mL, N-benzyl-3-piperidone (17.12g, 0.09mol), 2-chloro-3-bromopropene to a 500mL dry and clean reaction flask (22.66g, 0.135mol), a reflux condenser was installed, heated and stirred under reflux for 13 hours. Cool, add 20 mL of water, recover acetonitrile by distillation, add 100 mL of water, extract with ethyl acetate (100 mL×3), combine the ethyl acetate extracts, wash once with 50 mL of saturated saline, dry over anhydrous sodium sulfate, filter, and evaporate ethyl acetate The ester obtained 21.7 g of yellow viscous liquid, ie intermediate VIII, with a yield of 87%.
[0090] R f =0.6(petroleum ether:EtOAc=3:1); IR(KBr)v max : 2951, 1716, 1635, 1454, 1136, 885cm -1 ; 1 H NMR (400MHz, CDCl 3 ): δ1.97-2.04(m, 2H), 2.38-2.43(m, 1H), 2.45-2.57(m, 1H), 2.61-2.67(m, 1H), 2.84-2.86(m, 2H), 2.90 -2.95(m, 1H), 3.52(t, 1H, J=6.4Hz), 3.62(d, 1H, J...
Embodiment 3
[0091] Example 3 Preparation of intermediate VIII with another structure, the structural formula is as follows:
[0092]
[0093] Add anhydrous sodium carbonate (10.49g, 0.099mol), anhydrous sodium bromide (10.18g, 0.099mol), acetonitrile 200mL, N-benzyl-3-piperidone (17.12g , 0.09mol), 2-butenyl chloride (crotyl chloride, 11.78g, 0.130mol), a reflux condenser was installed, and the reaction was heated and stirred under reflux for 13 hours. Cool, add 20 mL of water, recover acetonitrile by distillation, add 100 mL of water, extract with ethyl acetate (100 mL×3), combine the ethyl acetate extracts, wash once with 50 mL of saturated saline, dry over anhydrous sodium sulfate, filter, and evaporate ethyl acetate The ester obtained 21.7 g of yellow viscous liquid, ie intermediate VIII, with a yield of 87%.
[0094] Rf = 0.7 (petroleum ether: EtOAc = 3: 1); 1H NMR (600MHz, CDCl3): δ0.89 (d, 3H, J = 6.0Hz, α), 1.11 (d, 3H, J = 6.0Hz, β ), 1.94-2.03(m, 2H, α+β), 2.23-2.39(m, 4H, ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More