Preparation method of dolutegravir ring-opening impurities, and impurities thereof
A dolutegravir and impurity technology, which is applied in the field of preparation of dolutegravir ring-opening impurities, can solve problems such as difficult separation and extraction, undisclosed preparation methods of impurity A and impurity B, and achieve good yield, easy research, and reaction conditions simple effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0049] refer to figure 1 , taking the preferred raw materials to synthesize dolutegravir ring-opening impurity A and impurity B as an example, the synthesis principle is as follows:
[0050] The first step: compound 1 and compound 2 react to generate compound 3
[0051] Using dichloromethane as a solvent and N,N-dicarbonylimidazole (CDI) as a condensation reagent, compound 1 and compound 2 undergo a condensation reaction at room temperature. Wash with saturated brine, evaporate the organic phase to dryness under reduced pressure, and refine Compound 3. The purification uses silica gel column chromatography, and the eluent uses n-hexane:ethyl acetate=3:1; the reaction equation is as follows:
[0052]
[0053] Step 2: Compound 3 is hydrolyzed to generate Compound 4
[0054] Stir and mix ethanol and compound 3, add 2N aqueous sodium hydroxide solution for hydrolysis reaction, adjust the pH to 3-4 with 6N aqueous hydrochloric acid solution at the end of the reaction, spin dry...
Embodiment 2
[0072] Compound 1 is condensed with compound 2 to generate compound 3:
[0073]
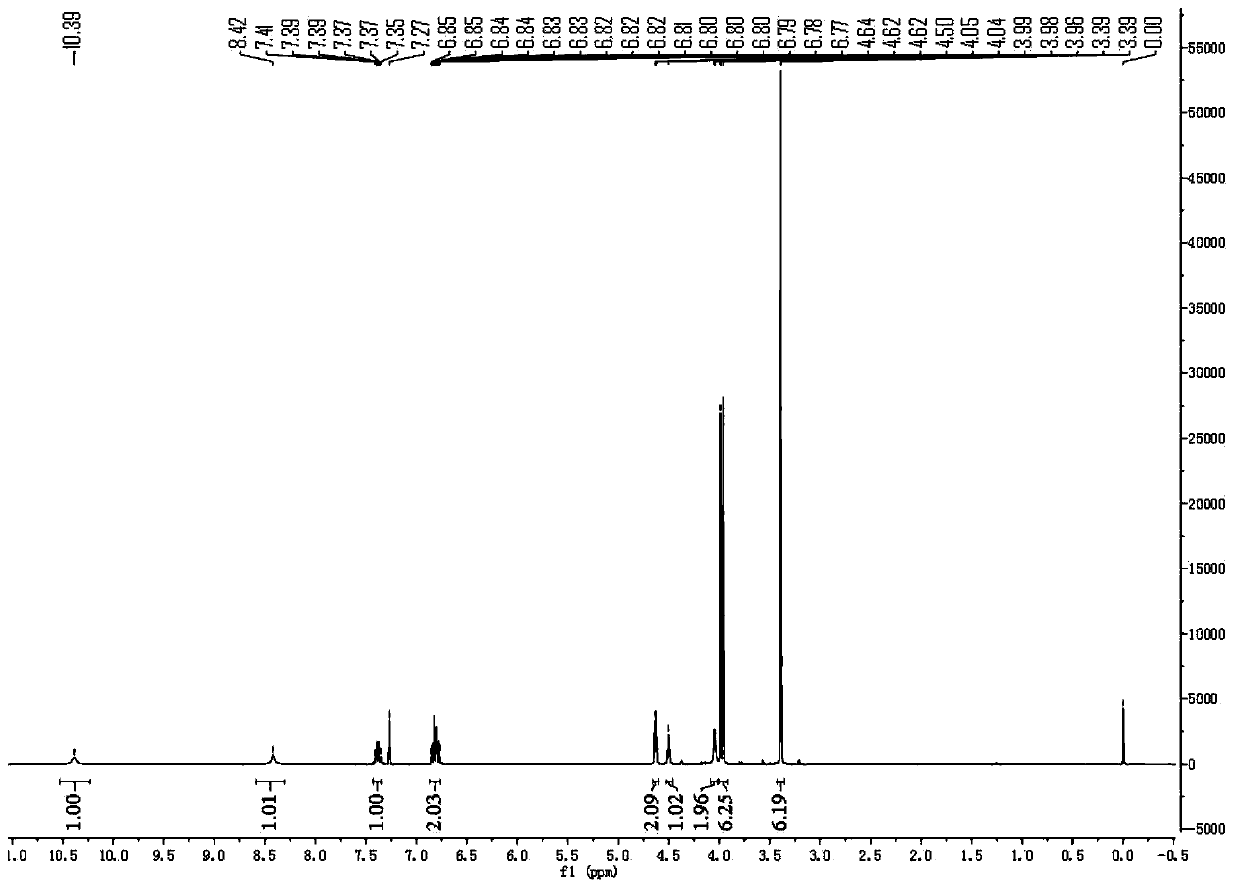
[0074] Compound 1 (20.0 g, 63.4 mmol) and dichloromethane (150 mL) were added into the reaction flask, and stirred at room temperature to dissolve. Cool down to 15-20°C, add N,N-carbonyldiimidazole (13.9 g, 85.7 mmol), and keep the reaction for 2 hours under the protection of nitrogen. Compound 2 (10.0 g, 69.9 mmol) was added dropwise, and the reaction was carried out at room temperature for 2 hours after the drop was completed. The reaction solution was washed successively with 1N aqueous HCl (100 mL), 5% aqueous sodium carbonate (130 mL), and saturated brine (100 mL). The organic phase was concentrated to dryness, and the obtained oil was separated by column chromatography [mobile phase: n-hexane: ethyl acetate (2:1)] to obtain compound 3 (18.9 g, 68%) as a white solid powder.
[0075] The proton nuclear magnetic spectrum of the obtained compound 3 sees figure 2 , and its H NMR and mass ...
Embodiment 3
[0077] Compound 3 prepared in Example 2 of the present invention was hydrolyzed under alkaline conditions to generate compound 4, and compound 4 was condensed with compound 5 to generate compound 6:
[0078]
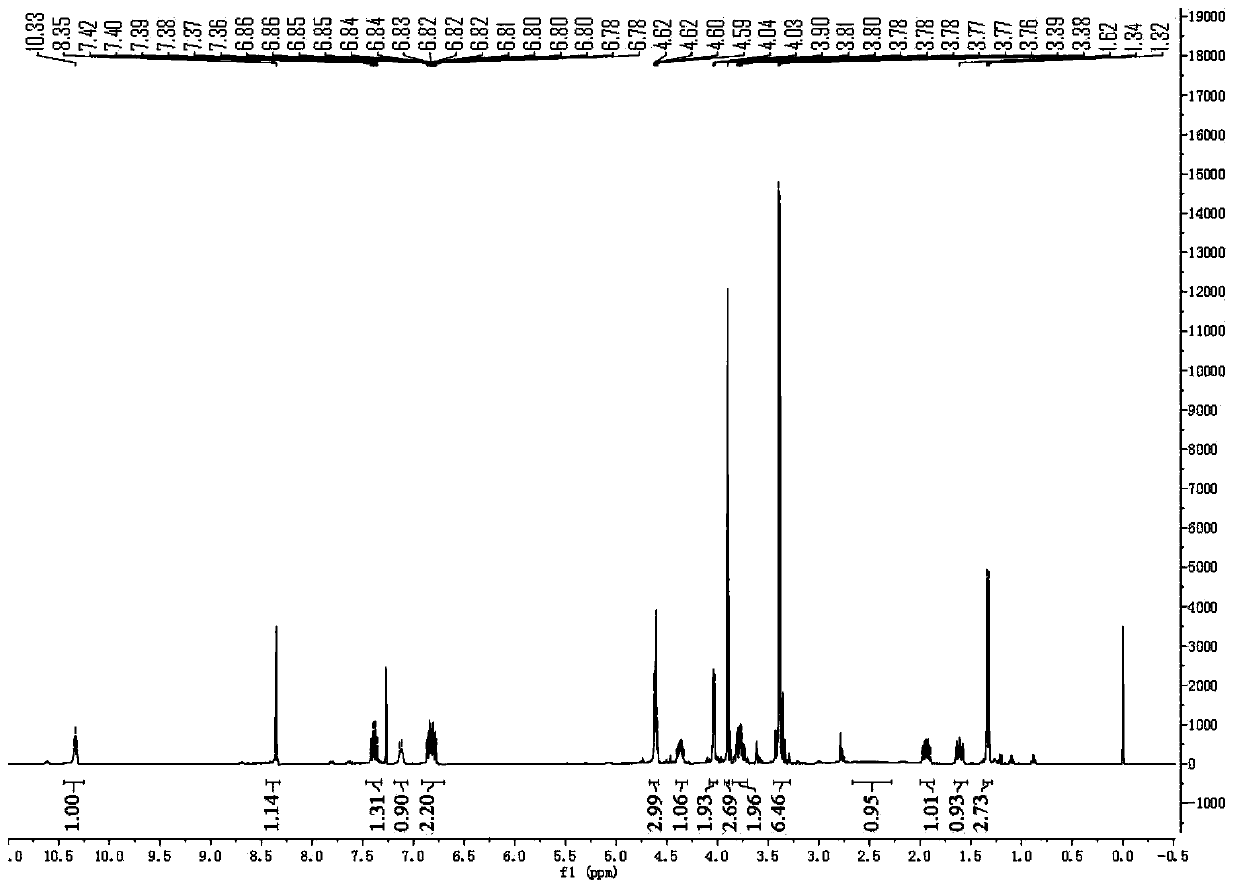
[0079] Add compound 3 (18.9 g, 42.9 mmol) and ethanol (190 mL) into the reaction flask, and stir to dissolve. 2N NaOH aqueous solution (95 mL) was added, and the reaction was stirred at room temperature for 1 hour, and TLC identified that the starting material disappeared. The pH was adjusted to 3-4 with 6N aqueous HCl. The reaction solution was concentrated to dryness, water (50 mL) was added to the residue, extracted with dichloromethane (100 mL×3), the combined organic phases were concentrated to dryness to obtain compound 4 as a white solid, which was directly added to dichloromethane (180 mL), and 1 -Hydroxybenzotriazole (8.7g, 64.4mmol), 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (12.3g, 64.4mmol), compound 5 (7.7g , 85.8 mmol). After stirring...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More