Enzyme preparation for removing o-nitrobenzoic acid (2-NBA) and application
A technology of o-nitrobenzoic acid and enzyme preparation, which is applied in the fields of enzyme genetic engineering and enzyme engineering, can solve problems such as the reduction of decomposition rate of organic matter, and achieve the effects of excellent effect, mild reaction conditions and high enzyme activity
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

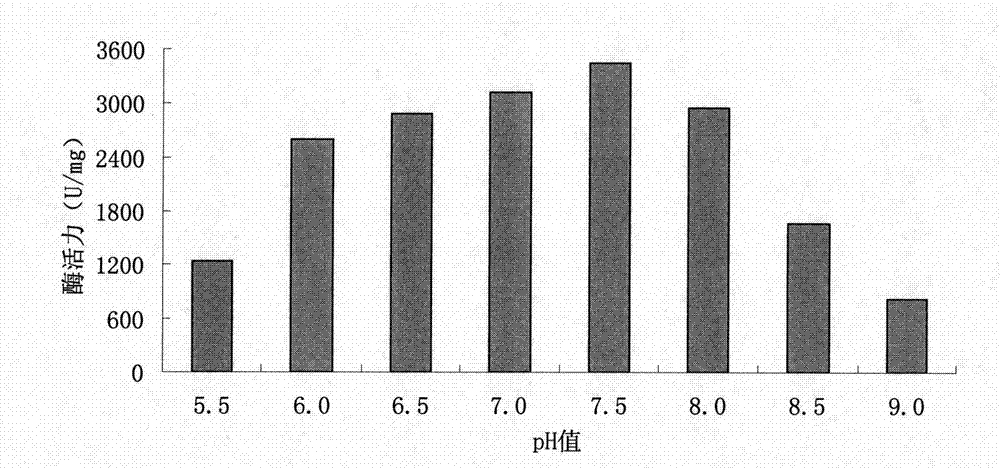
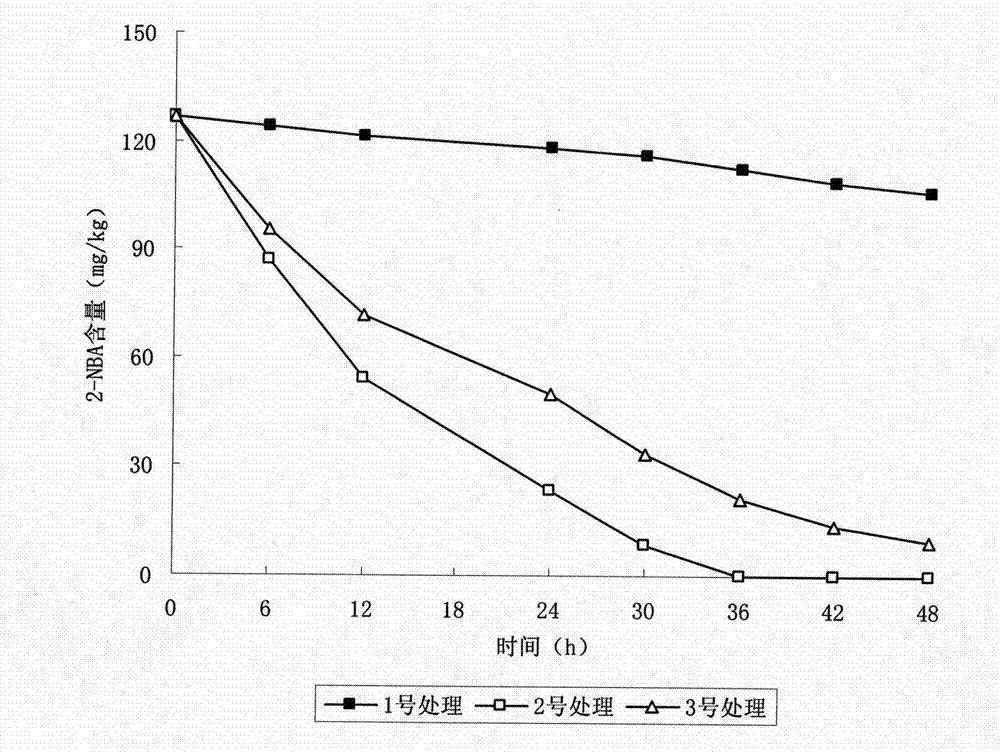
Examples
Embodiment 1
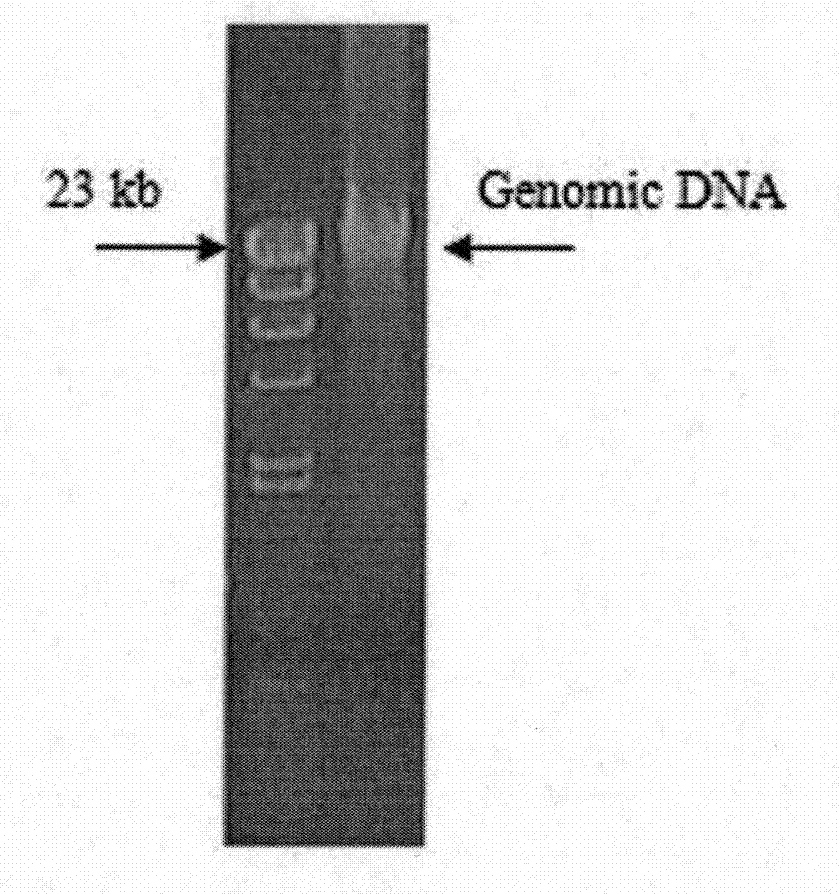
[0023] The extraction of embodiment 1Pseudomonas sp.ONBA-17 total DNA (Genomic DNA)
[0024] 1.1 Expanded cultivation of Pseudomonas sp.ONBA-17
[0025] Under a sterile operating environment, small pieces of Pseudomonas sp.ONBA-17 stored at -70°C were picked with an inoculation needle, placed and spread on Luria-Bertani (LB) solid medium. 28 ℃, static culture 3d. A single colony was selected, and after two transfers, it was observed that the colonies on the plate had the same shape (no bacteria). The "plate" and Luria-Bertani (LB) solid / liquid medium described here are all common terms, medium, techniques and methods in the field of microbial research / production. For details, see "Molecular Cloning Experiment Guide" (J. Sam Brook, et al. Molecular Cloning Experiment Guide (Third Edition) [M]. Huang Peitang, et al. Translated. Beijing: Science Press, 2008). Unless otherwise specified, the techniques, methods, culture media, reagents and medicines used in microbial research a...
Embodiment 22
[0029] The acquisition of embodiment 22-NBA degrading enzyme coding gene nbaB
[0030] After a large number of analysis experiments, a pair of effective amplification primers (denoted as SEQ ID No.3 and SEQ ID No.4 respectively) were screened out from 24 pairs of self-designed primers.
[0031] nbaB-f,5'-ACGAC CATATG AGTTACCAAAACTTAG-3' (the underline is the NdeI restriction site, and the gene sequence is SEQ ID No.3);
[0032] nbaB-r, 5'-CATCA GAATTC GGAAGACCAGGAGC-3' (the underline is the EcoRI restriction site, and the gene sequence is SEQ ID No.4).
[0033] 25 μL amplification system: 2.5 μL of 10×Taq DNA polymerase reaction buffer; 2 μL of dNTP (25 mM); 0.5 μL of each primer (25 pmol / μL); Mg 2+ Buffer (25mM) 2.5μL; total DNA obtained in Example 1 0.5μL (about 100ng); Taq enzyme (5U / μL) 0.3μL; ddH 2 016.2 μL. Reaction parameters: pre-denaturation at 95°C for 5 min, denaturation at 94°C for 30 sec, annealing at 52°C for 30 sec, extension at 72°C for 1 min, amplificat...
Embodiment 3
[0037] Construction, expression and purification of embodiment 3 expression vector
[0038] The amplified product of the nbaB gene in Example 2 was digested with Nde I and EcoRI, and then the fragment was inserted into the pET21b(+) vector digested with the same restriction endonuclease. In the recombinant plasmid, the nbaB gene is driven by the T7 promoter, the C-terminus of the expression product has 6 histidine tags, and ampicillin is the selection marker. The joined fragments were verified by Shanghai Sangon sequencing to ensure that no mutations were introduced during the PCR process.
[0039] The recombinant plasmid was transferred into Escherichia coli BL21 (DE3), and then the strain containing the recombinant plasmid (numbered Y-6) was selected, and the foreign gene introduction was verified by Shanghai Sangon sequencing. Y-6 was cultured with LB based on 37°C culture, after A 600nm When the value was 0.6, IPTG was added to a final concentration of 1 mM, followed by ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - Generate Ideas
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com