

Sitagliptin synthesis method
A synthetic method and step-by-step technology, applied in the field of medicine and chemical industry, can solve the problems of resource waste, many steps, expensive raw materials, etc., and achieve the effects of avoiding toxic and dangerous chemical reagents, mild reaction conditions, and easy-to-obtain raw materials
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

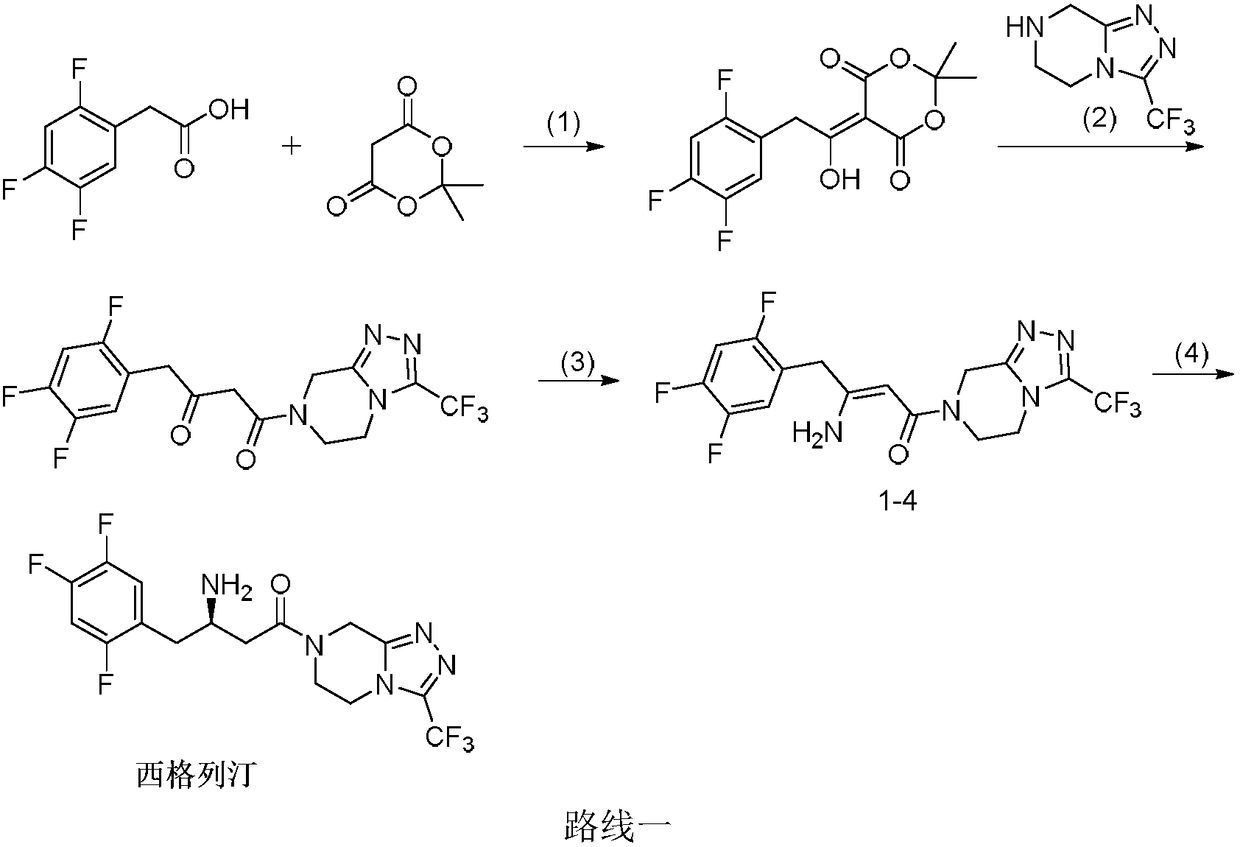
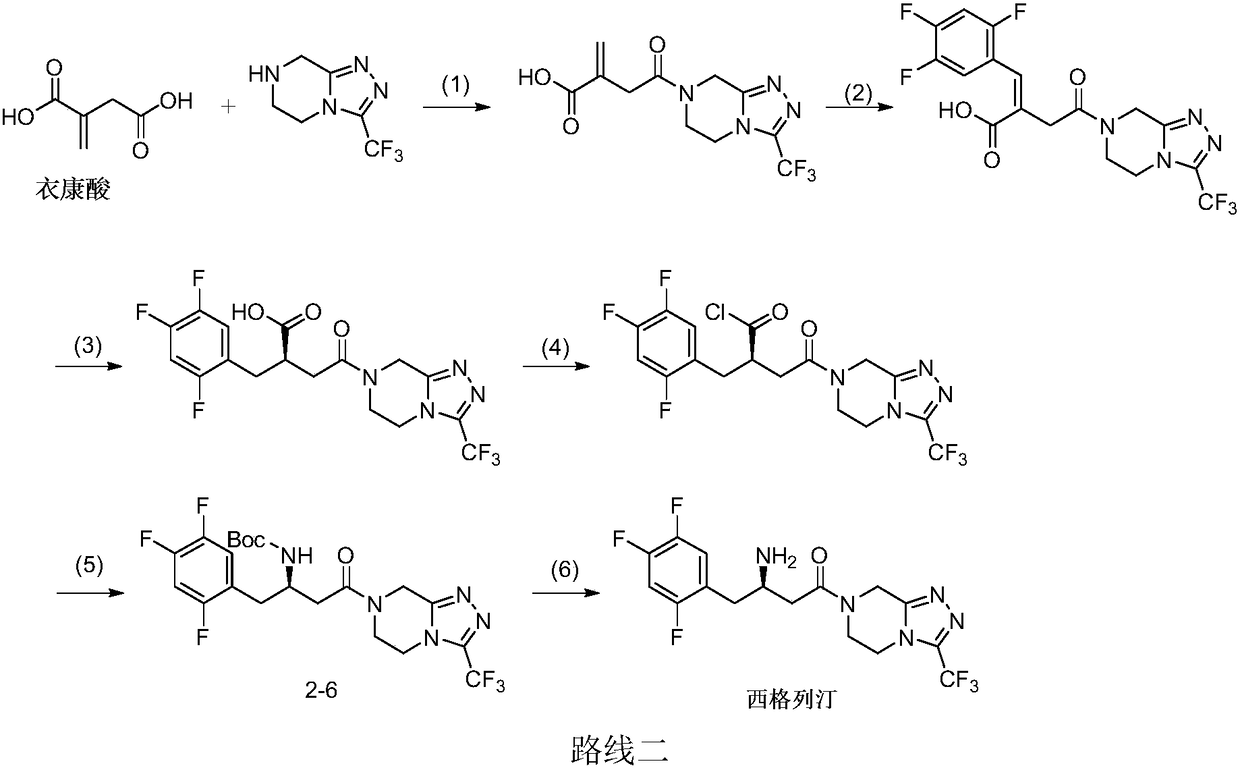
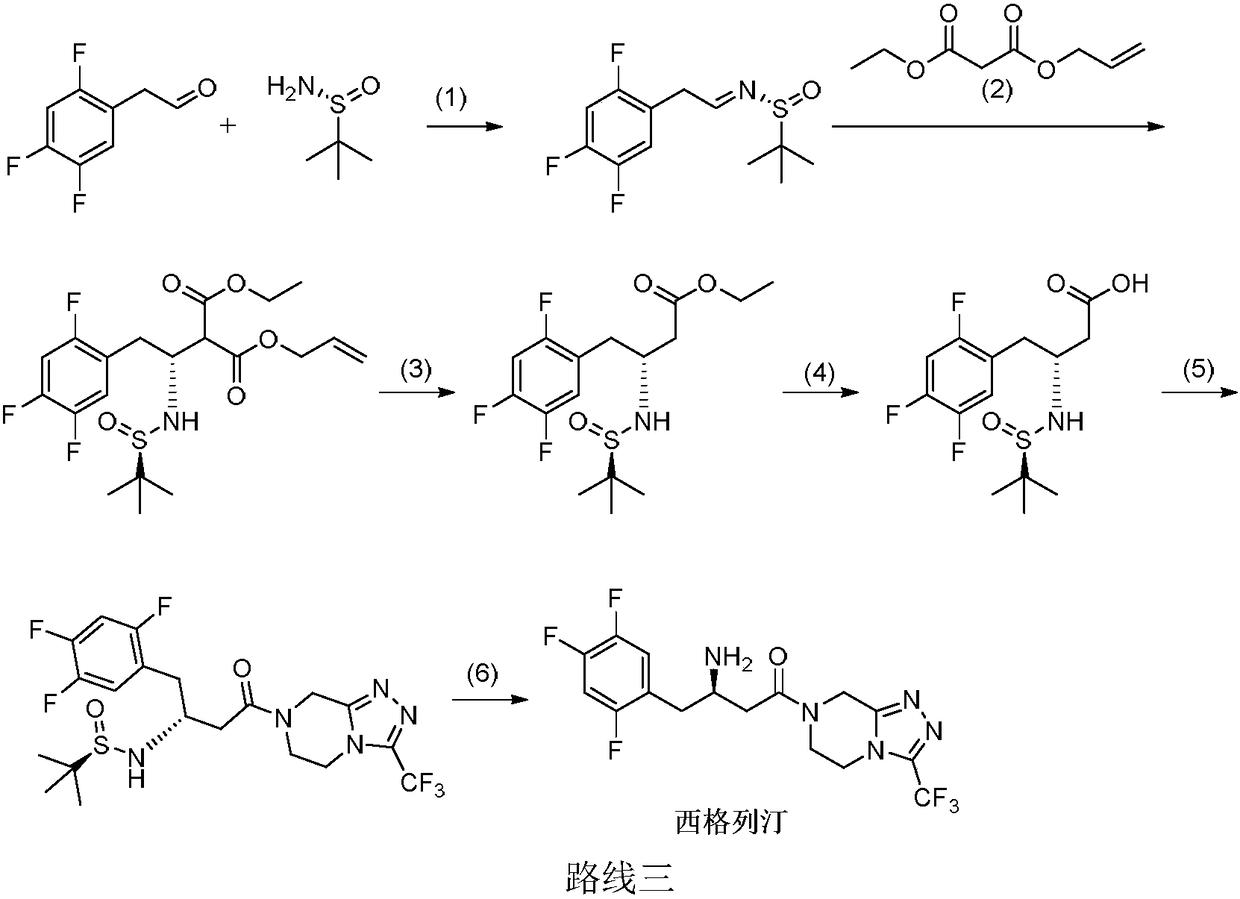
Examples
Embodiment 1
[0034] Embodiment 1, the preparation of formula II compound
[0035] In a 50mL single-necked bottle, add 2,4,5-trifluorobromobenzene (I) (2.10g, 10mmol), diethyl malonate (4.80g, 30mmol), cuprous iodide (0.19g, 1mmol), L-proline (0.23g, 2mmol), potassium phosphate (7.43g, 35mmol) and 20mL of anhydrous DMSO, and the reaction system was replaced with nitrogen three times. Raise the temperature to 90°C and react for 24h. After that, spread a layer of diatomaceous earth on the filter paper, filter, and wash the filter cake with 10 mL of ethyl acetate to obtain the mother liquor. Add 70 mL of ethyl acetate to dilute the mother liquor, wash three times with saturated ammonium chloride aqueous solution, once with saturated sodium chloride aqueous solution, and finally wash once with water, and spin evaporate the ethyl acetate under reduced pressure to obtain the crude product. The crude product was recrystallized from methanol at 0° C. to obtain 2.28 g of a light yellow solid, name...
Embodiment 2
[0036] Embodiment 2, the preparation of formula III compound
[0037] Into a 50 mL single-necked flask, 1-tert-butoxycarbonylpiperazine (1.87 g, 10 mmol), trifluoroacetic anhydride (3.15 g, 15 mmol) and 15 mL of anhydrous dioxane were sequentially added, and stirred at room temperature for one hour. Then hydrazine hydrate (80%, 1.25g, 20mmol), tetrabutylammonium iodide (0.37g, 1mmol) and morpholine (0.17g, 2mmol) were added successively, and then the temperature was raised to 70°C. Hydrogen peroxide (30%, 4.53g, 40mmol) was dissolved in 5mL of dioxane, and the hydrogen peroxide solution was slowly added dropwise to the above reaction solution for 4h, and then the reaction was continued for 2h. Afterwards, 80 mL of ethyl acetate was added to dilute the mother liquor, washed three times with saturated aqueous sodium sulfite solution, once with saturated aqueous sodium chloride solution, and finally washed once with water, and the ethyl acetate was rotary evaporated under reduced p...
Embodiment 3
[0038] Embodiment 3, the preparation of formula IV compound
[0039] In a 100 mL single-necked bottle, potassium tert-butoxide (2.24 g, 20 mmol) was dissolved in 40 mL of tetrahydrofuran, and stirred at room temperature for one minute. Then compound III (1.91 g, 10 mmol) and compound II (2.60 g, 10 mmol) were added sequentially, and stirring was continued at room temperature for 60 min. Afterwards, the reaction solvent tetrahydrofuran was distilled off under reduced pressure, and 50 mL of water and 50 mL of dichloromethane were added to extract an organic phase. The organic phase was dried with anhydrous sodium sulfate, and then distilled under reduced pressure to obtain a yellow crude product solid. The crude product was recrystallized from dichloromethane and cyclohexane to obtain 3.33 g of compound IV with a yield of 82% and a purity of 99%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More