A kind of preparation method of Mirabegron
An intermediate and reaction technology, applied in the field of preparation of Mirabegron, can solve problems such as difficulty in realizing industrial production, difficulty in obtaining starting materials, long synthetic route, etc., and achieve fewer steps in the synthetic route, broad prospects and industrial application Good effect of value, purity
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

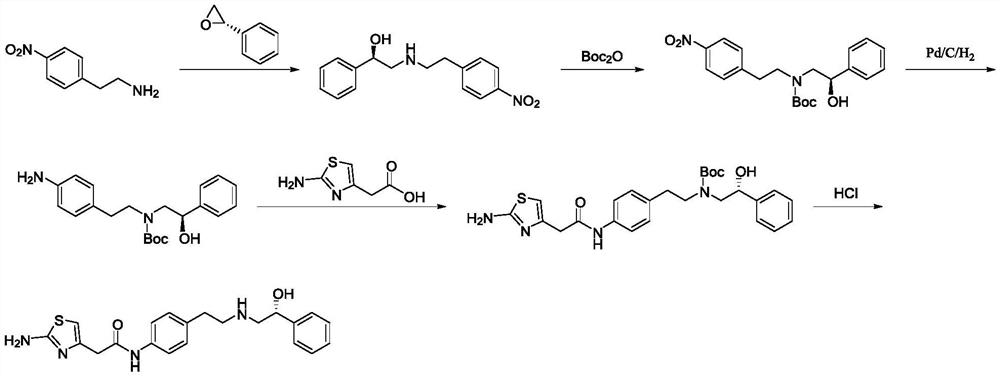

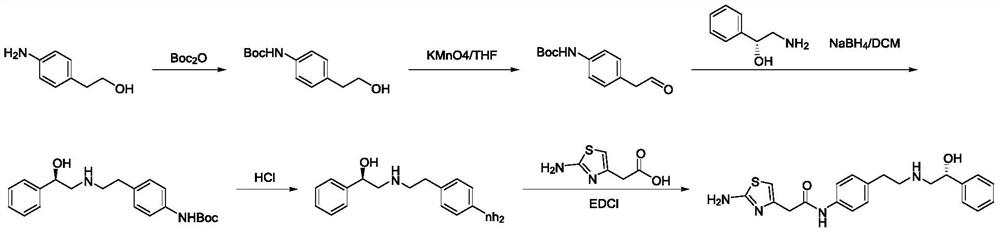
Examples
Embodiment 1
[0042] Synthesis of S1, intermediate (R)-2-hydroxyl-N-(4-nitrophenethyl)-2-phenylacetamide
[0043] Add 273g (1.64mol, 1.0eq) of p-nitrophenylethylamine, (R)-mandelic acid 250g (1.64mol, 1.0eq) and 1500mL of xylene (mandelic acid V / m is 6), under the protection of nitrogen, the temperature is raised to 140 ° C for reflux, and the reaction is kept for 7 hours. The progress of the reaction is monitored by HPLC. Dissolve methane, wash with 500mL*2 of 5% dilute hydrochloric acid and 500mL*2 of 5% sodium hydroxide respectively, dry the organic phase over anhydrous sodium sulfate, remove dichloromethane under reduced pressure, and obtain the product after drying (R)-2-Hydroxy-N-(4-nitrophenethyl)-2-phenylacetamide is about 429 g, the yield is 87.2%, and the purity is 99.2%.
[0044] Synthesis of S2, intermediate (R)-2-((4-nitrophenethyl)amino)-1-phenylethanol hydrochloride
[0045]Add 400g (1.33mol, 1.0eq) of the intermediate (R)-2-hydroxy-N-(4-nitrophenethyl)-2-phenylacetamide, ...
Embodiment 2
[0051] Synthesis of S1, intermediate (R)-2-hydroxyl-N-(4-nitrophenethyl)-2-phenylacetamide
[0052] Add 273g (1.64mol, 1.0eq) of p-nitrophenylethylamine, (R)-mandelic acid 499g (3.28mol, 2.0eq) and 1750mL of xylene (mandelic acid V / m is 3.5), under the protection of nitrogen, the temperature is raised to 160°C and refluxed, and the reaction is kept for 10 hours. The progress of the reaction is monitored by HPLC. Dissolve methane, wash with 500mL*2 of 5% dilute hydrochloric acid and 500mL*2 of 5% sodium hydroxide respectively, dry the organic phase over anhydrous sodium sulfate, remove dichloromethane under reduced pressure, and obtain the product after drying (R)-2-Hydroxy-N-(4-nitrophenethyl)-2-phenylacetamide is about 450 g, the yield is 91.5%, and the purity is 99.3%.
[0053] Synthesis of S2, intermediate (R)-2-((4-nitrophenethyl)amino)-1-phenylethanol hydrochloride
[0054] Add 400g (1.33mol, 1.0eq) of the intermediate (R)-2-hydroxy-N-(4-nitrophenethyl)-2-phenylacetami...
Embodiment 3
[0060] Synthesis of S1, intermediate (R)-2-hydroxyl-N-(4-nitrophenethyl)-2-phenylacetamide
[0061] Add 273g (1.64mol, 1.0eq) of p-nitrophenylethylamine, (R)-mandelic acid 375g (2.46mol, 1.5eq) and 1875mL of dimethylbenzene (mandelic acid V / m is 5), under the protection of nitrogen, the temperature is raised to 150 ° C and refluxed, and the reaction is kept for 9 hours. The progress of the reaction is monitored by HPLC. Dissolve methane, wash with 500mL*2 of 5% dilute hydrochloric acid and 500mL*2 of 5% sodium hydroxide respectively, dry the organic phase over anhydrous sodium sulfate, remove dichloromethane under reduced pressure, and obtain the product after drying (R)-2-Hydroxy-N-(4-nitrophenethyl)-2-phenylacetamide is about 454 g, the yield is 92.3%, and the purity is 99.2%.
[0062] Synthesis of S2, intermediate (R)-2-((4-nitrophenethyl)amino)-1-phenylethanol hydrochloride
[0063] Add 400g (1.33mol, 1.0eq) of the intermediate (R)-2-hydroxy-N-(4-nitrophenethyl)-2-pheny...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com