

Method for identifying BPDE-added target protein
An identification method and target protein technology, applied in measuring devices, instruments, scientific instruments, etc., can solve problems such as lack, impossibility of chemical modification, discount of protein information, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment
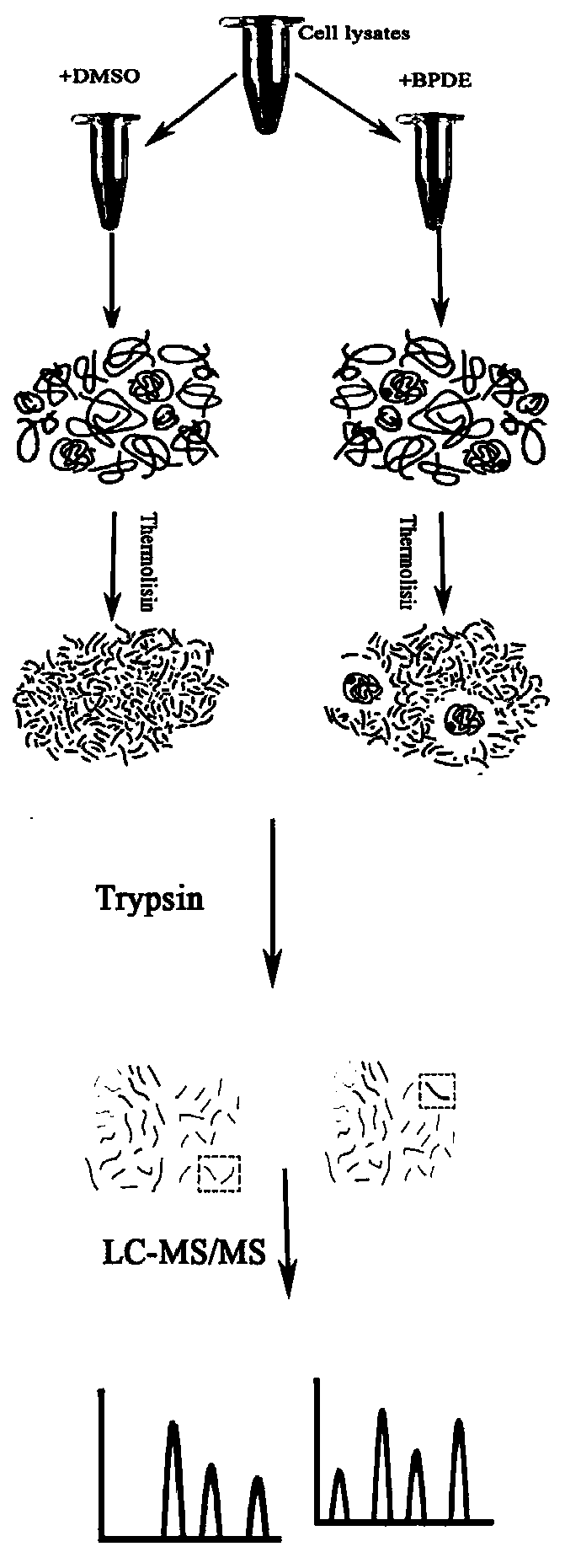
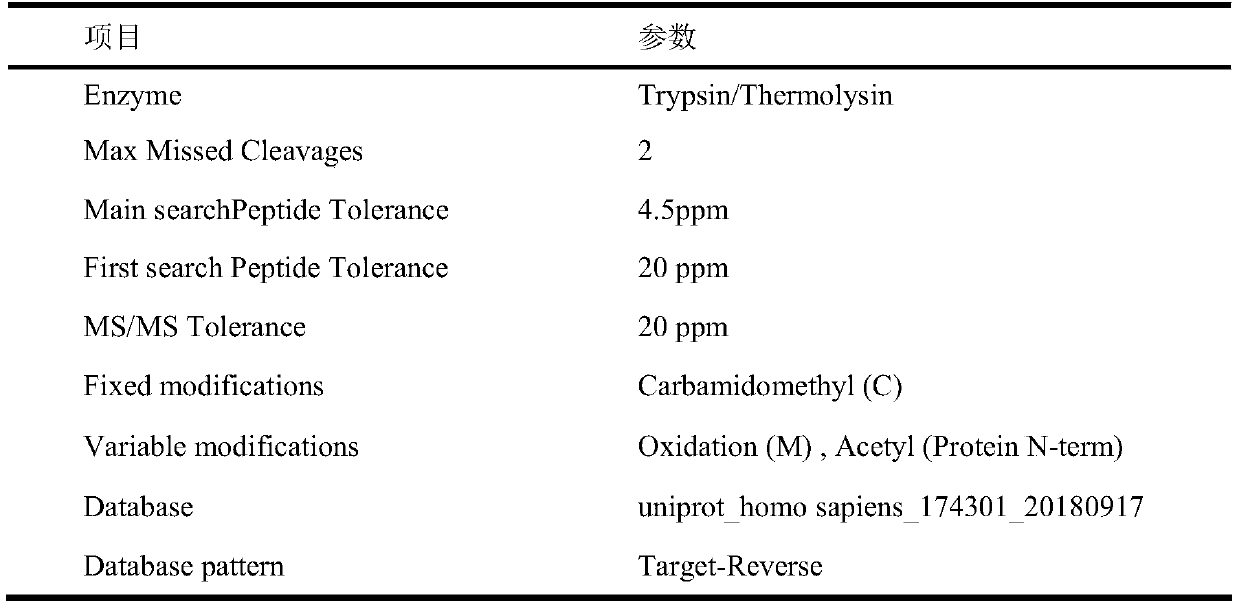
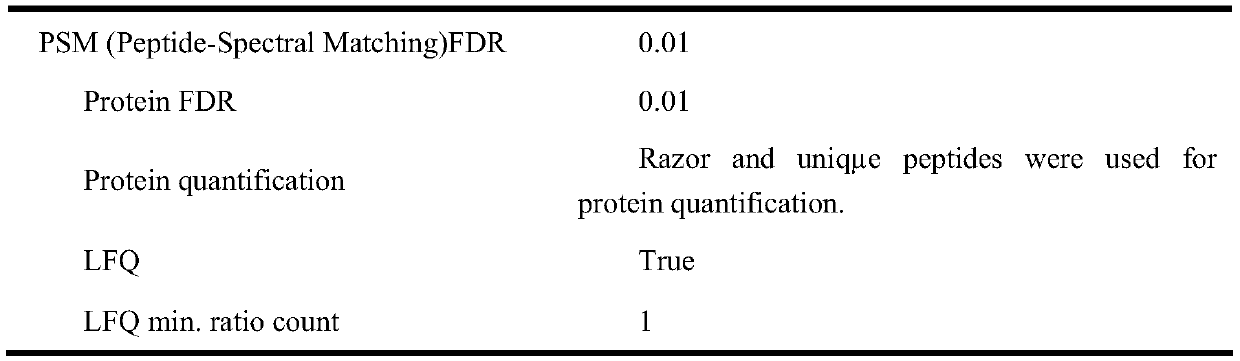
[0041] The identification method of BPDE addition target protein in human neuroma blastoma cell (SH-SY5Y) lysate provided by the embodiment (method process see figure 1 ) and results, including the following steps:
[0042] Step 1, SH-SY5Y cell culture and protein extraction
[0043] Neuroblastoma cell line SH-SY5Y was incubated at 37°C, 5% CO 2 Cultivate under certain conditions, subculture (2-3 days / time), and when the cell confluence reaches 80% to 85%, discard the medium, wash the cells twice with 5 mL pre-cooled PBS buffer, add trypsin to digest and collect the cells, Centrifuge at 1000rpm for 5min, resuspend in 5mL of pre-cooled PBS buffer, discard the supernatant after centrifugation (repeat cycle to wash cells 3 times), add 100μL of pre-cooled PBS buffer to suspend cells, freeze in liquid nitrogen for 1min, take out Shake and thaw in a water bath at 25°C (repeat this cycle of freezing and thawing 4 times), centrifuge at 20,000×g (acceleration of gravity) and 4°C for ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More