Method for preparing faropenem daloxate
A technology for faropenem axetil and ropenem axetil is applied in the field of preparation of faropenem axetil, and can solve the problems of inability to prepare faropenem axetil, high price of tetrabutylammonium fluoride, unattainable yield and quality, and the like. And the effect of high purity of finished product, lower cost and improved quality of finished product
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
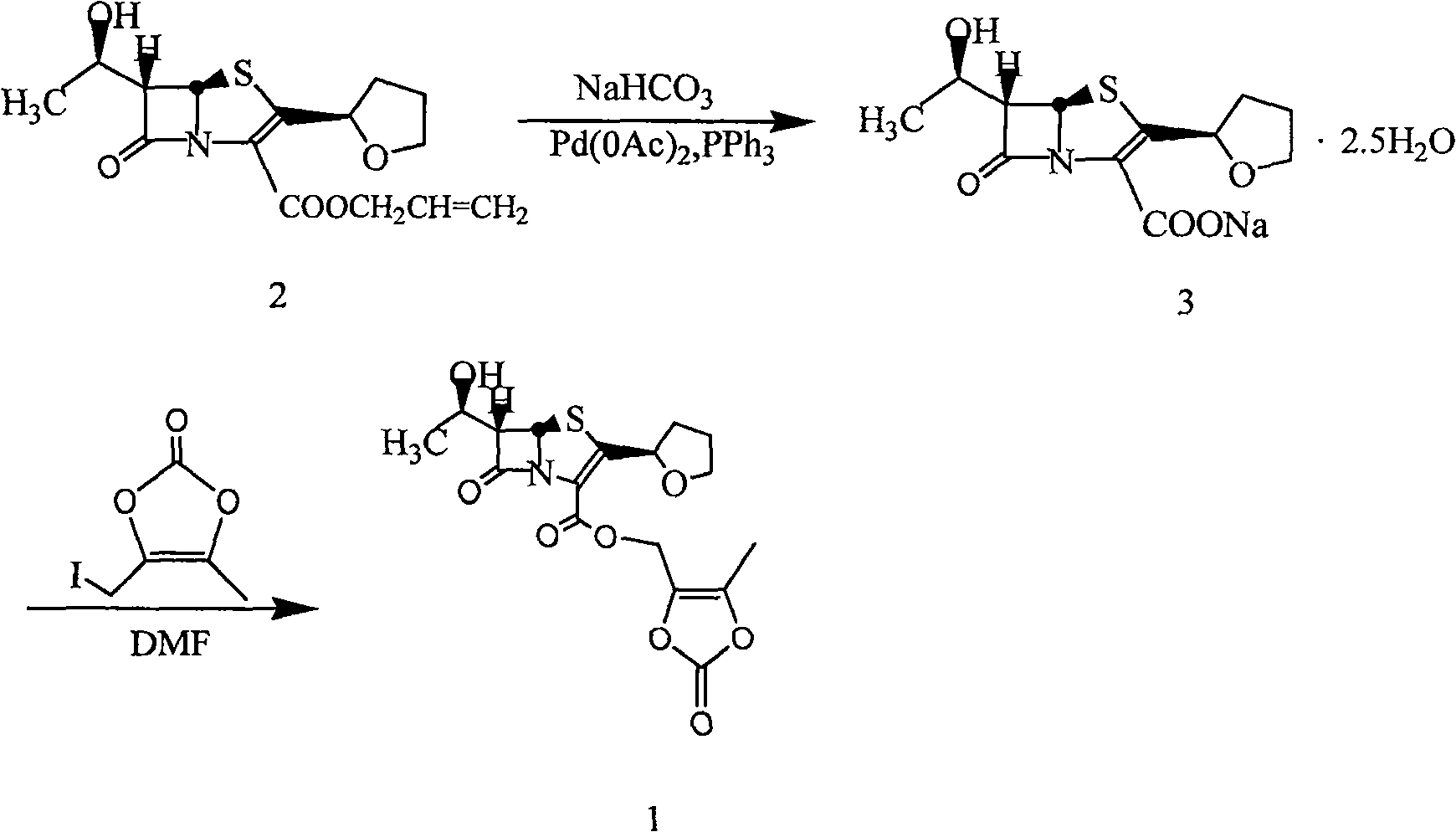
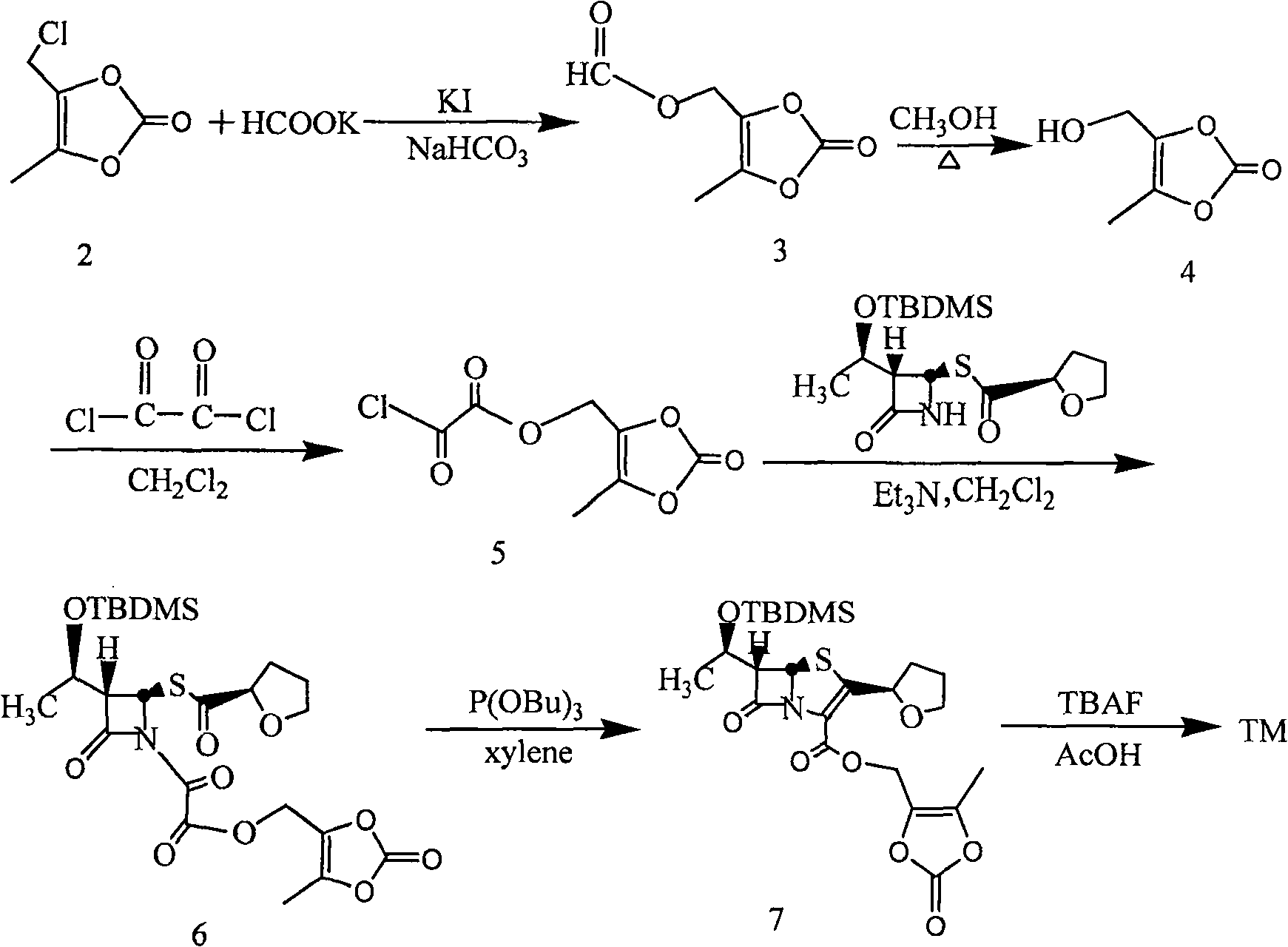
[0047] Add 570g of faropenem and 2500mL of 1,3-dimethylpropylene urea into a 10000mL three-necked bottle, stir and dissolve at room temperature, then add 260g of K 2 CO 3 , 50g 18-crown-6, 70g KI, stirred for 1 hr, cooled to 0~5°C, added dropwise 320g 4-methyl-5-chloromethyl-1,3-dioxol-2-one (DMDO-Cl), and control the reaction temperature between 0 and 5°C. After dropping, stir at the same temperature for 6hr. Add 2500 mL of ice water and 3000 mL of ethyl acetate and stir for 20 minutes, let stand, separate the organic layer, wash with 2500 mL of saturated brine, and dry over anhydrous magnesium sulfate. The desiccant was removed by filtration, and the filtrate was concentrated to dryness under reduced pressure at 40°C. The residue was recrystallized from a mixed solution of ethyl acetate:n-hexane=2:1 to obtain 687g of faropenem ester, with a yield of 86.5%, mp: 122-125°C, [M+1] + : 398.41.
Embodiment 2
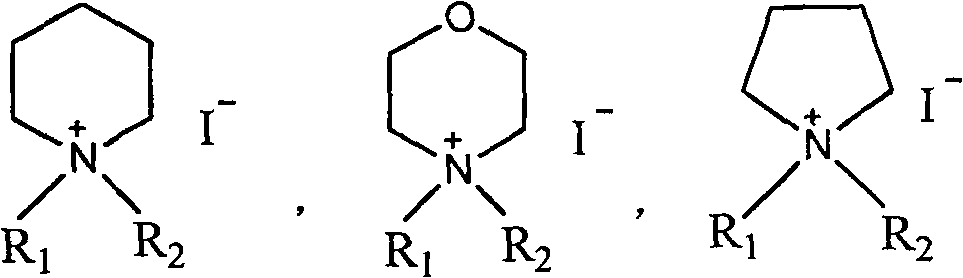
[0049] Add 50g of N-methylmorpholine, 500mL of dichloromethane, and 100mL of iodomethane into a 1000mL three-necked flask, heat up and reflux for 3 hours, then concentrate under reduced pressure to dryness to obtain a yellow N, N-dimethylmorpholine iodide solid, which is added to Add 4000mL of acetone to a 10000mL three-necked flask, add 570g of faropenem and 50g of 18-crown-6 under stirring, stir at room temperature for 30 minutes, and adjust the pH to 7.5-8.5 with N-methylmorpholine. Cool in an ice bath to 0-5°C, add 320g of DMDO-Cl dropwise, and control the reaction temperature between 0-5°C. After dropping, stir at the same temperature for 6hr. Add 3000mL of water, stir and crystallize at 0~5℃ for 4hr. The solid was collected by filtration. Vacuum-dried at 40°C to obtain the crude yellow faropenem ester, which was recrystallized with 75% aqueous methanol to obtain 670g of the finished product with a yield of 84.4%, mp: 123-125.5°C, [M+1] + : 398.41.
Embodiment 3
[0051] Add 57g of faropenem and 250mL of dimethyl sulfoxide into a 1000mL three-necked bottle, stir and dissolve at room temperature, then add 32g of NaHCO 3 , 5g 18-crown-6, 7g sodium iodide, after stirring for 3hr, under cooling, dropwise add 32g (4-methyl-5-chloromethyl-1,3-dioxol-2-one ( DMDO-Cl), control the reaction temperature between 20~25°C. After dropping, stir at the same temperature for 6hr. Add 250mL of ice water and 300mL of ethyl acetate and stir for 20 minutes, let stand, separate the organic layer, 250mL of saturated saline Wash and dry over anhydrous magnesium sulfate. Remove the desiccant by filtration, and concentrate the filtrate to dryness under reduced pressure at 40°C. The residue is recrystallized from a mixed solution of ethyl acetate:n-hexane=2:1 to obtain 66g of faropenem ester, with a yield of 83.1 %, mp: 122~125℃, [M+1] + : 398.41.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More