Recombinant plasmid containing OATP1B1 promoter and reporter gene and method for screening OATP1B1 inducer
A reporter gene and recombinant plasmid technology, applied in the field of plasmids containing fluorescent reporter genes, can solve the problems of rarely used, human hepatocyte model ethics, and tissue source culture conditions can not achieve high throughput, etc., to achieve easy materials, The effect of fast detection speed and high sensitivity
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1
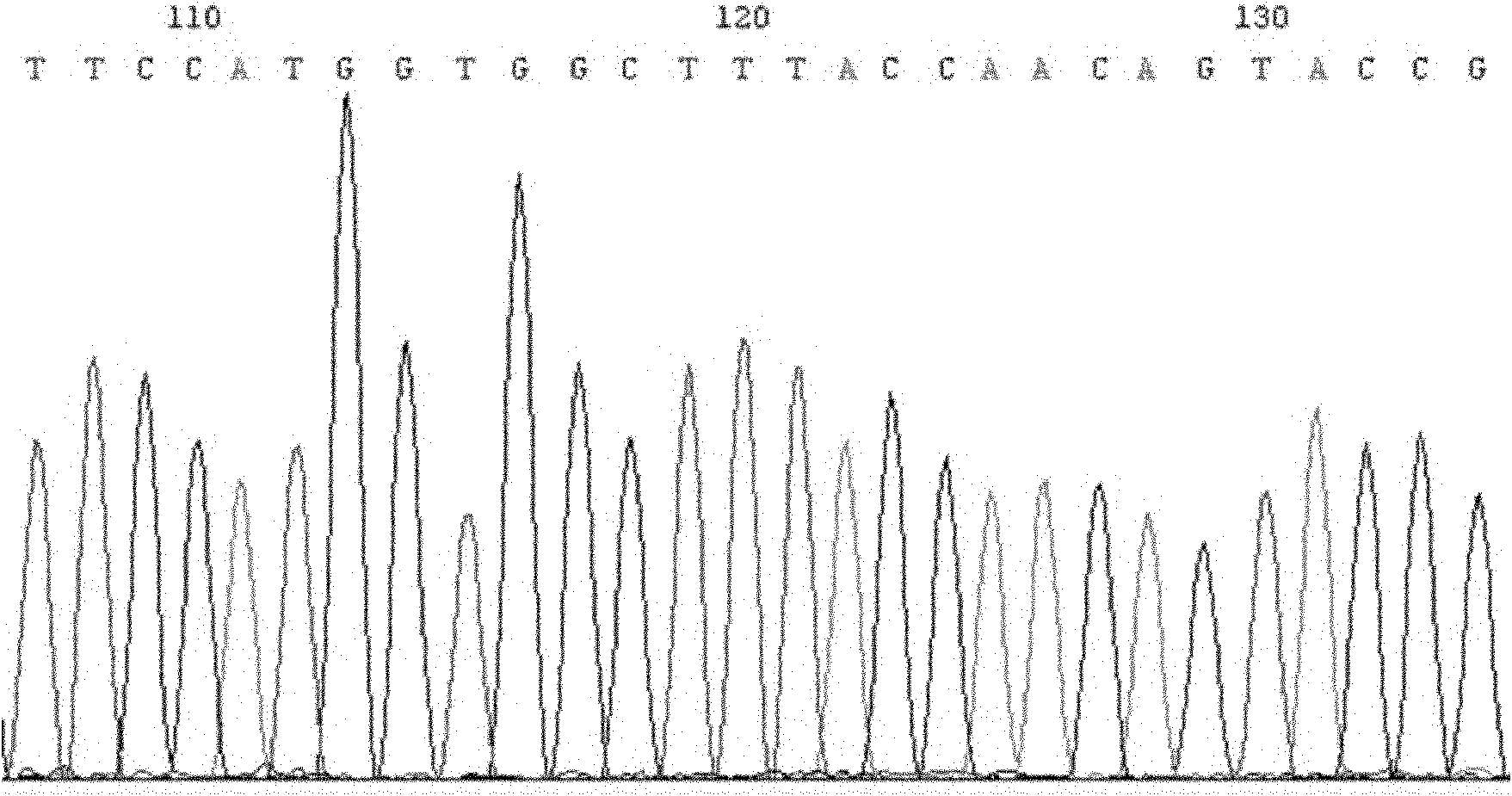
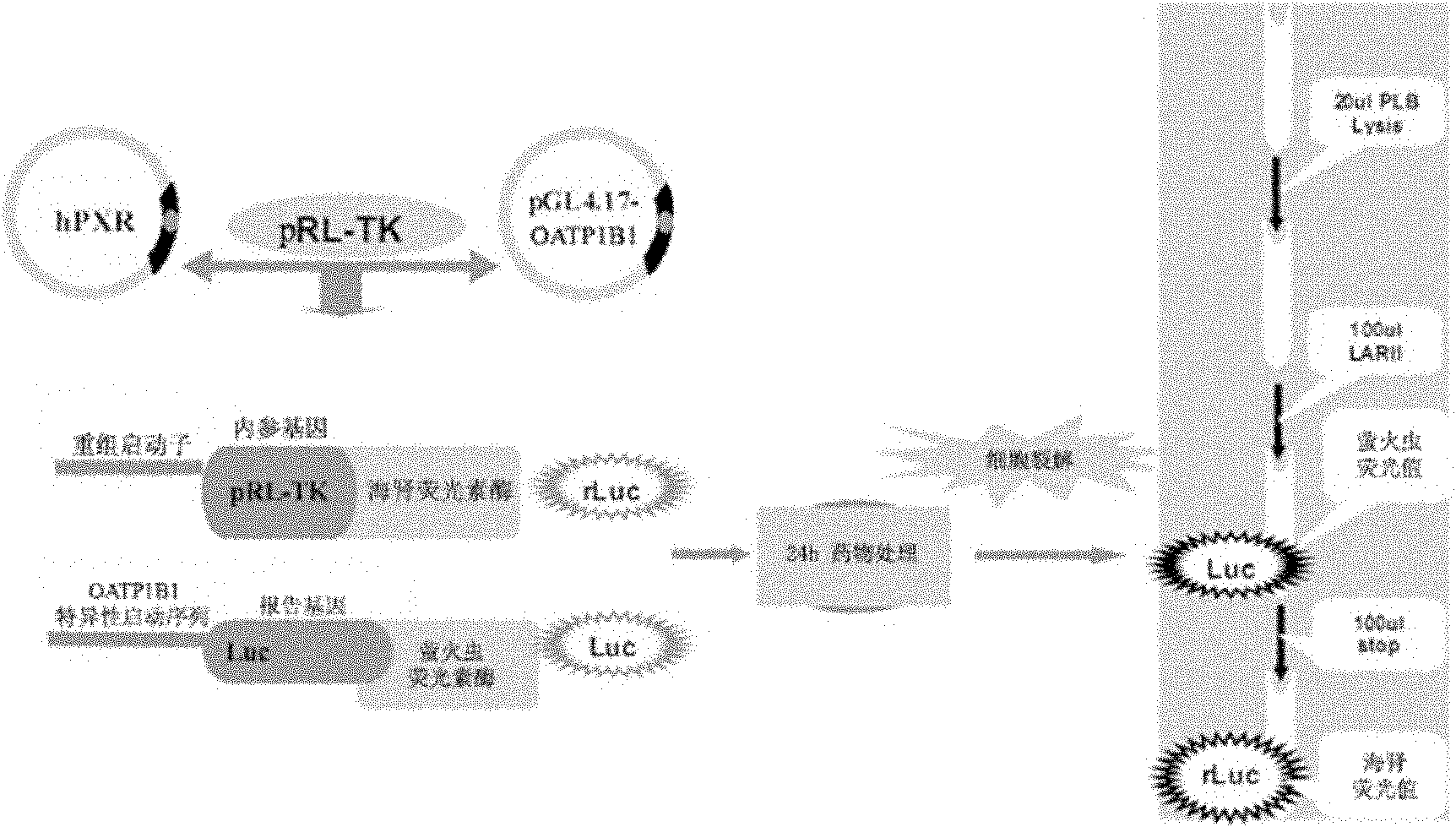
[0030] The OATP1B1 promoter fluorescent reporter gene plasmid was constructed, and transiently transfected into HepG2 cells to construct a dual fluorescent reporter gene technology platform.
[0031] 1. Materials
[0033] pGL-4.17 fluorescent reporter gene vector, pPEM-T vector, pcDNA3.1-myc / hisB(-) eukaryotic expression vector: American Promega Company.
[0034] HepG2 cell line: Tumor Cell Bank, Chinese Academy of Medical Sciences.
[0035] 2. Main reagents
[0036] Fetal bovine serum (Hangzhou Sijiqing); MEM medium (Gibco, USA); 50ml cell culture flask, 24-well cell culture plate (Corning, USA); RevertAid TM First Strand cDNA Reverse Transcription Kit (Fermentas); Lipofectamine TM 2000 liposome (Invitrogen, USA); DNA extraction kit (promega); Trizol (Invitrogen, USA).
[0037] 3. Main instruments
[0038] PCR instrument (Thermo, USA); electrophoresis equipment (Beijing Liuyi); CO 2 Cell incubator (ThermoForm, Series II, USA); ultra-cle...
Embodiment 2
[0158] The experimental method established above was used to study whether the traditional Chinese medicine monomer compound baicalin could induce the expression of OATP1B1 by activating PXR.
[0159] Steps:
[0160] 1. Culture HepG2 cells;
[0161] When HepG2 cells grow to the logarithmic phase, digest with 2×10 per well 5 Cells were seeded into 24-well plates until the cells grew to 80%.
[0162] 2. Co-transfection
[0163] 2.1 Each well needs to be transferred into 600ng PXR plasmid, 300ng OATP1B1 promoter fluorescent reporter gene plasmid and 50ng sea cucumber luciferase respectively. Dilute the DNA and Lipofectamine2000 liposomes with an appropriate amount of culture medium without antibiotics and fetal bovine serum, mix the two, and place them at room temperature for 20 minutes.
[0164] 2.2 Wash the cells three times with PBS, and then add 1ml of MEM medium without fetal bovine serum to each well.
[0165] 2.3 Add the above mixture (DNA / liposome) into a 24-well pla...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More