

Cefcapene pivoxil hydrochloride and method for preparing its intermediate
A technology of cefcapene pivoxil and cefcapene acid, which is applied in the field of preparation of cefcapene pivoxil hydrochloride and its intermediates, can solve the problems of long reaction steps and difficult availability of starting materials, and achieve few synthesis steps and cheap raw materials , the effect of simple operation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

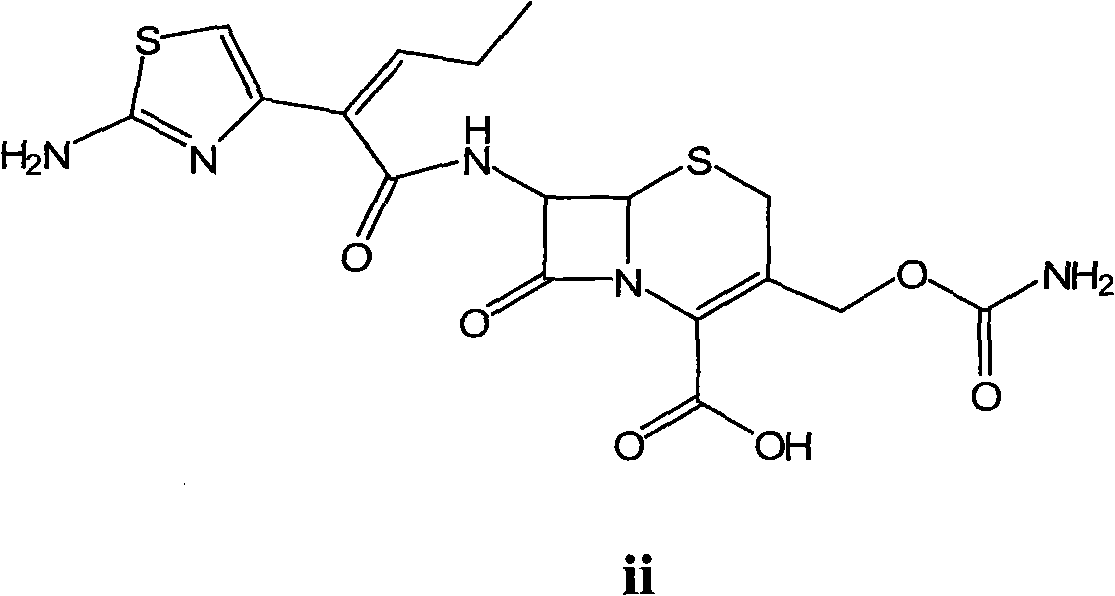
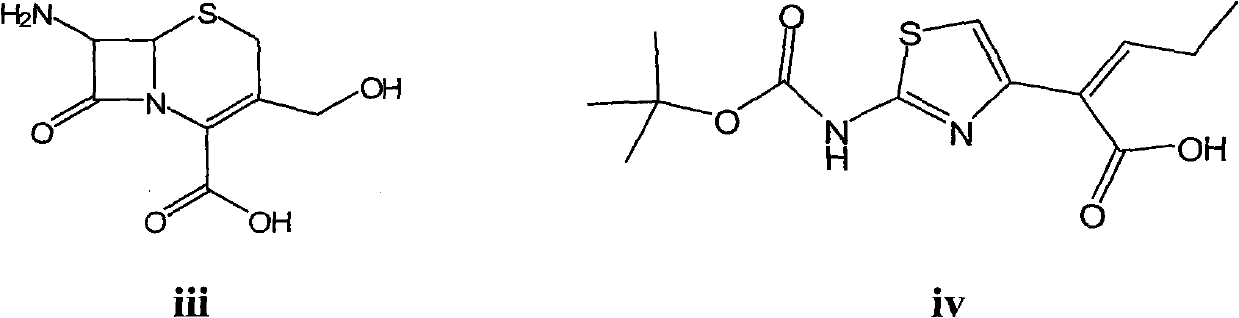
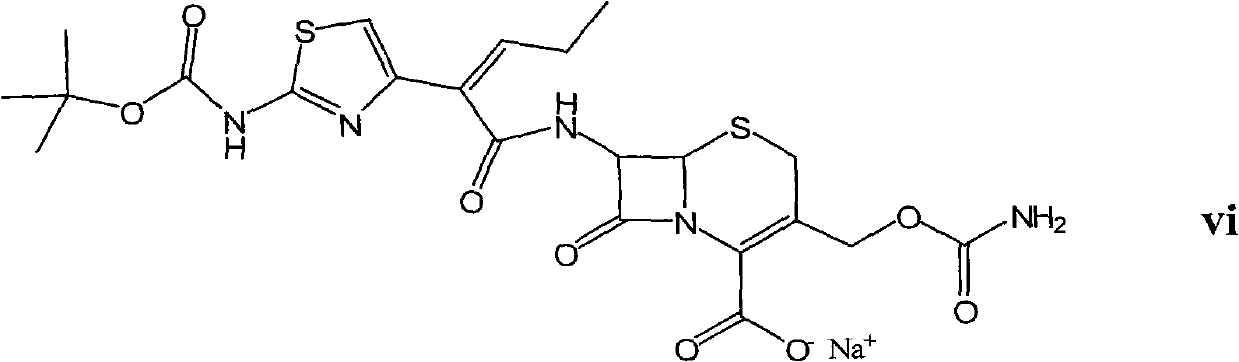
Examples
Embodiment 1
[0050] Embodiment 1: Preparation of tert-butoxycarbonyl cefcapene sodium salt (compound of formula vi)
[0051] Add 7-DACA (100 g, 0.43 mol), 500 mL of dichloromethane, and 55 ml of tetramethylguanidine into a 1 L reaction flask, and stir to dissolve. Cool down to -20°C and keep warm to prepare for the following condensation reaction.
[0052] Add cefcapine side-chain acid (130 g, 0.44 mol), 1.3 L of dichloromethane, and 90 ml of triethylamine into a 5 L reaction flask, and stir to dissolve. The temperature was lowered to -20°C, 60ml of methanesulfonyl chloride was added, and the reaction was stirred for 30min. Then add the prepared 7-DACA feed solution, and react at -20°C for 3 hours. Then chlorosulfonyl isocyanate (42ml, 0.48mol) was added, and the reaction was carried out at -20°C for 2 hours. Add 800 mL of tetrahydrofuran and water, 100 g of sodium bicarbonate, and stir for 30 min. The phases were separated, and the organic phase was washed once with water, and a sodium ...
Embodiment 2
[0053] Embodiment 2: Preparation of tert-butoxycarbonyl cefcapene sodium salt (compound of formula vi)
[0054] Add 7-DACA (100 g, 0.43 mol), 500 mL of dichloromethane, and 55 ml of tetramethylguanidine into a 1 L reaction flask, and stir to dissolve. Cool down to -15°C and keep warm to prepare for the following condensation reaction.
[0055] Add cefcapine side-chain acid (137 g, 0.46 mol), 1.3 L of dichloromethane, and 96 ml of diisopropylamine into a 5 L reaction flask, and stir to dissolve. Cool down to -15°C, add 67ml of methanesulfonyl chloride, stir for 30 minutes, add the prepared 7-DACA feed solution, and react at -15°C for 3 hours. Then trichloroacetyl isocyanate (63.2ml, 0.53mol) was added, and the temperature was controlled at -15°C for 2 hours. Add 800 mL each of tetrahydrofuran and water, add 139 g of sodium carbonate, and stir for 30 minutes. The phases were separated, the organic phase was washed once with water, a solution of sodium isooctanoate in tetrahyd...
Embodiment 3
[0056] Embodiment 3: the preparation of cefcapene acid (formula ii compound)
[0057] Add tert-butoxycarbonyl cefcapene sodium salt (100 g, 0.17 mol), 600 mL of methanol, and 600 mL of 1N HCl into a 2L reaction flask, and stir and react at -10°C for 8 hours. After the reaction was completed, the pH of the solution was adjusted to 7.5 with saturated sodium bicarbonate, and extracted with ethyl acetate. The resulting aqueous phase was adjusted to pH 3 with 3N HCl, and a solid was precipitated, filtered, washed, and dried to obtain 69.5 g of the title compound as a white solid, with a molar yield of 87% and an HPLC purity of 99.2%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More