

A new synthetic method of JAK inhibitor baricitinib and its intermediate
A technology for baricitinib and a synthesis method, applied in the field of medicine and chemical industry, can solve the problems of expensive 3-hydroxyazetidine starting material, unsatisfactory product yield and purity, poor acetonitrile stability, etc. Route efficiency and atom economy, simplified separation and purification process, simple operation effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

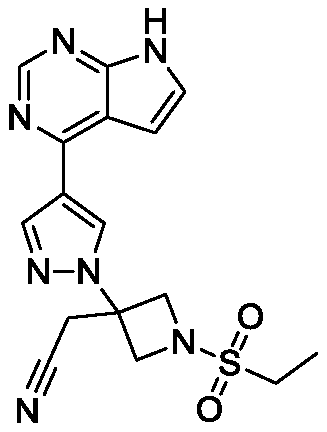
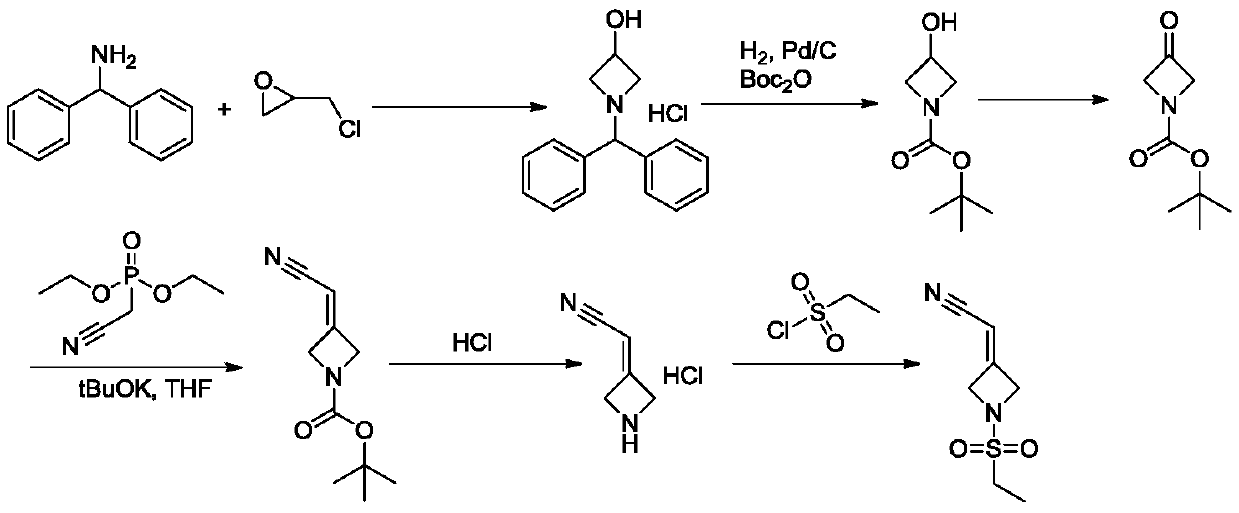
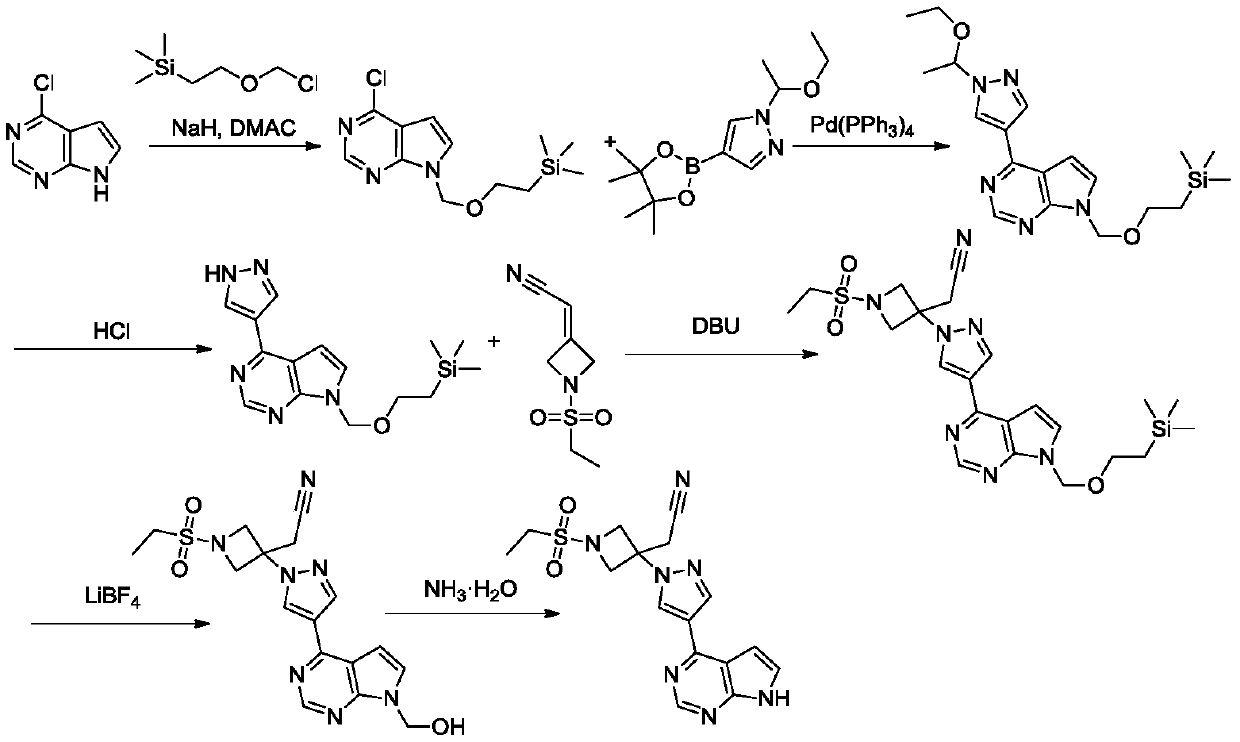
Examples
Embodiment 1
[0047] Example 1: N-(3-chloro-2-hydroxypropyl)ethanesulfonamide
[0048]
[0049] Add 1-amino-3-chloropropyl-2-ol hydrochloride (14.60g, 100mmol), tetrahydrofuran (73mL), water (73mL) into a three-necked flask, stir to dissolve and cool to 0~5℃, add hydrogen phosphate Dipotassium (34.84g, 200mmol), stir for 5 minutes and then add ethyl sulfonyl chloride (13.50g, 105mmol) dropwise. After the addition, warm up to room temperature and react for 3-4 hours. At the end of the reaction, add 73mL of 0.5mol / L dilute hydrochloric acid to quench Reaction, stirring and liquid separation, the aqueous phase was extracted twice with 35 mL ethyl acetate, the combined organic phase was washed once with saturated brine (73 mL), dried over anhydrous sodium sulfate, and concentrated to obtain N-(3-chloro-2-hydroxypropyl) The crude product of ethyl sulfonamide is directly applied to the next reaction (GC purity is about 92%).
[0050] ESI m / z=202.1(M+H) + , 1 H NMR(400MHz, CDCl 3 )δ5.10-4.95(m,1H), 4....
Embodiment 2
[0052] Example 2: 1-(Ethylsulfonyl)azidine-3-ol
[0053]
[0054] Add N-(3-chloro-2-hydroxypropyl)ethanesulfonamide (prepared from Example 1) and N,N-dimethylformamide (100mL) into a three-necked flask, stir to dissolve, and cool to 0~5℃. Potassium tert-butoxide (11.22g, 100mmol) was added, after the addition, kept at 0~5℃ and stirred for 15~20 minutes, then warmed to room temperature and reacted for 3-4 hours. Add 1mol / L dilute hydrochloric acid 100mL to quench the reaction, stir and separate. The aqueous phase was extracted twice with 50 mL ethyl acetate, and the combined organic phases were washed once with saturated brine (100 mL), dried over anhydrous sodium sulfate, and concentrated to obtain crude 1-(ethylsulfonyl)azidine-3-ol directly Throw into the next reaction (GC purity about 86%). ESI m / z=166.3(M+H) + .
[0055] In Example 2, the alkaline substance potassium tert-butoxide can be sodium hydride, sodium tert-butoxide, lithium diisopropylamide, lithium hexamethyldisilaz...
Embodiment 3
[0056] Example 3: 1-(Ethylsulfonyl)azidine-3-one
[0057]
[0058] A three-necked flask was charged with 1-(ethylsulfonyl)azidine-3-ol (prepared from Example 2), sodium bromide (10.29g, 100mmol), sodium bicarbonate (12.6g, 150mmol), dichloromethane (83mL) and water (123mL), stir well and then cool to -5~0℃, add TEMPO (312mg, 2mmol), add 10% sodium hypochlorite solution (81.9g, 110mmol) dropwise after the addition, the reaction is over and liquid separation, water The phase was extracted with dichloromethane (83mL) once again, the combined organic phase was washed with sodium bisulfite solution (5%, 42mL) once, washed with saturated brine once (83mL), dried over sodium sulfate, and concentrated to obtain 1-( The crude ethylsulfonyl)azidine-3-one (GC purity about 83%) was directly used in the next reaction. ESI m / z=164.0(M+H) +
[0059] The additives sodium bromide and sodium bicarbonate in Example 3 can be replaced by one or more combinations of triethylamine, diisopropylethylamin...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More