

Novel synthetic method for tofacitinib intermediate (3R,4R)-N,4-dimethyl-1-benzyl-3-piperidinamine and oxalate hydrates of (3R,4R)-N,4-dimethyl-1-benzyl-3-piperidinamine
A technology of tofacitinib and intermediates, applied in the field of medicinal chemistry, can solve the problems of poor stereoselectivity, poor atom economy, and low overall yield, and achieve high reaction yield, high optical purity, and cheap and easy-to-obtain raw materials Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

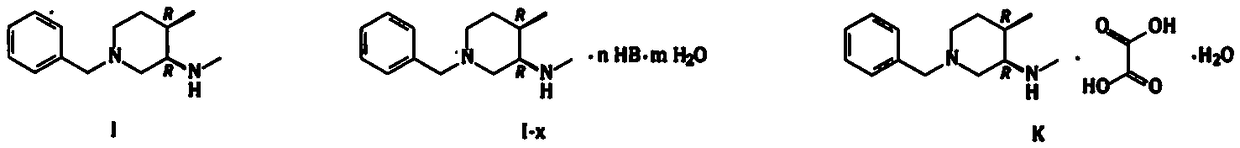
Examples
Embodiment 1
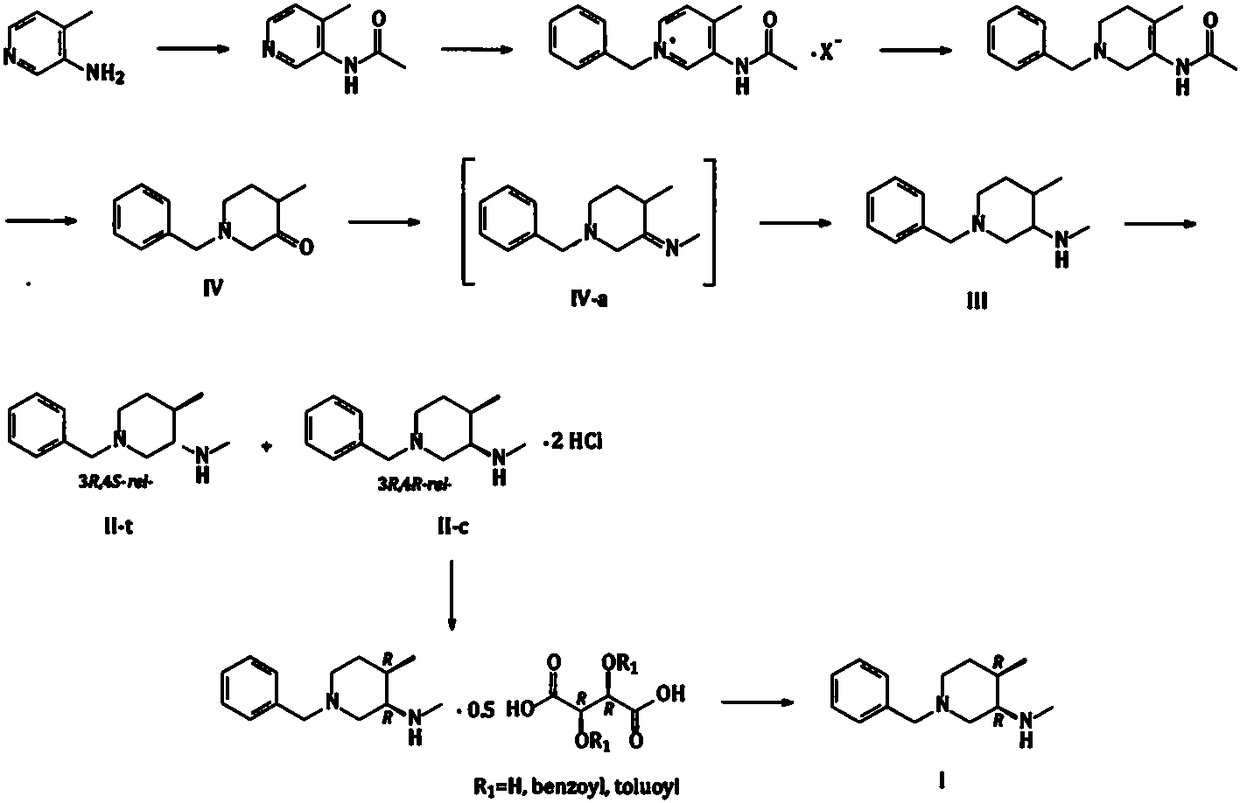
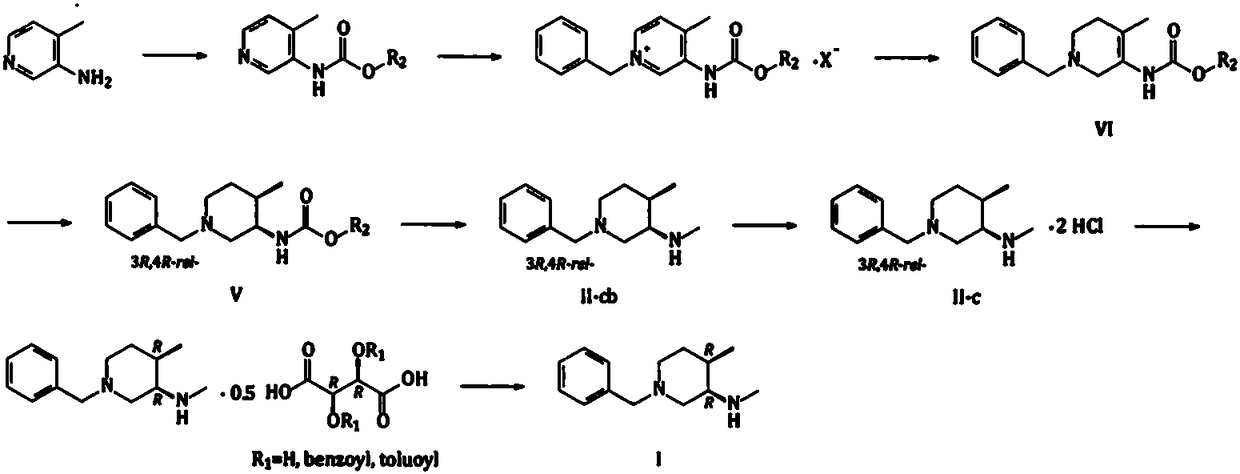
[0048] Embodiment 1: N-Boc-N, the synthesis of 4-dimethyl-3-aminopyridine (compound C)
[0049] Add 3-amino-4-picoline (100g, 924.7mmol) into the reaction flask, then add triethyl orthoformate (400g, 2.699mol), heat to 130-145°C for about 8-12hrs. After the reaction was completed, the temperature was lowered to about 100°C, and the triethyl orthoformate was concentrated under reduced pressure. The obtained residue was distilled under reduced pressure with a high vacuum oil pump to obtain 129 g (785.6 mmol) of light yellow to colorless liquid (Compound A-x, R=Et), with a yield of 85.0%.
[0050] The compound A-x (R=Et) (120g, 730.8mmol) obtained above was dissolved in 600g of absolute ethanol, cooled to between 5 and 10°C, and 32g (845.9mmol) of sodium borohydride was added in batches to control the temperature not to exceed 20°C After the addition, return to 25-30°C and stir for about 30-60 minutes, then slowly heat to reflux, and continue the reaction for about 2-4 hours. A...
Embodiment 2
[0052] Embodiment 2: N-Boc-N, the synthesis of 4-dimethyl-3-aminopyridine (compound C)
[0053] Add 3-amino-4-picoline (120 g, 1.110 mol) to the reaction flask, then add trimethyl orthoformate (450 g, 3.036 mol), and heat to 120-130°C for about 10-16 hrs. After the reaction was completed, the temperature was lowered to about 100°C, and the trimethyl orthoformate was concentrated under reduced pressure. The resulting residue (compound A-x, R=Me) (182.2 g based on theoretical calculation) was directly used in the next reaction.
[0054] Dissolve the compound A-x (R=Me) obtained above in 1080g of isopropanol, cool to 5-10°C, add 45g (1.190mol) of sodium borohydride in batches to control the temperature not to exceed 20°C, and return to Warm to 25-30°C and stir for about 1-1.5 hours, then slowly heat to reflux, and continue the reaction for about 2-4 hours. After the reaction, the isopropanol was concentrated under reduced pressure, water (1000g) was added to the residue, and TH...
Embodiment 3
[0056] Example 3: Synthesis of 1-benzyl-N-Boc-N,4-dimethyl-1,2,5,6-tetrahydropyridin-3-amine (compound E)
[0057] Add the obtained compound C (150g, 674.8mmol) to 600g toluene, then add benzyl chloride (94g, 742.6mmol), heat to 100-110°C, and react for 10-16hr; after the reaction is completed, cool to 20- 30°C, add 600g of water, stir evenly, let stand, separate the liquid, and separate the lower aqueous phase (aqueous solution of compound D-x, X - = Cl - ),spare.
[0058] Dissolve 0.1g of sodium hydroxide in 100g of water, then add sodium borohydride (26g, 687.3mmol) to dissolve in the aqueous sodium hydroxide solution; slowly add the aqueous solution of sodium borohydride dropwise to the above aqueous solution of quaternary ammonium salt, add dropwise The process temperature is controlled between 15 and 25°C. After the dropwise addition is completed, continue the heat preservation reaction for 2 to 4 hours; after the reaction is completed, add ethyl acetate for extraction...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More