

Silicon-center chiral silicon-oxygen compound and preparation method thereof
A technology of silicon oxide compounds and compounds, applied in the direction of silicon organic compounds, chemical instruments and methods, compounds of group 4/14 elements of the periodic table, etc., can solve the limitation of chiral silicon compound design and application, and cannot obtain satisfactory ee value , limited range of substrates, etc., to achieve the effects of wide application range of substrates, excellent glum value, and atom economy
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

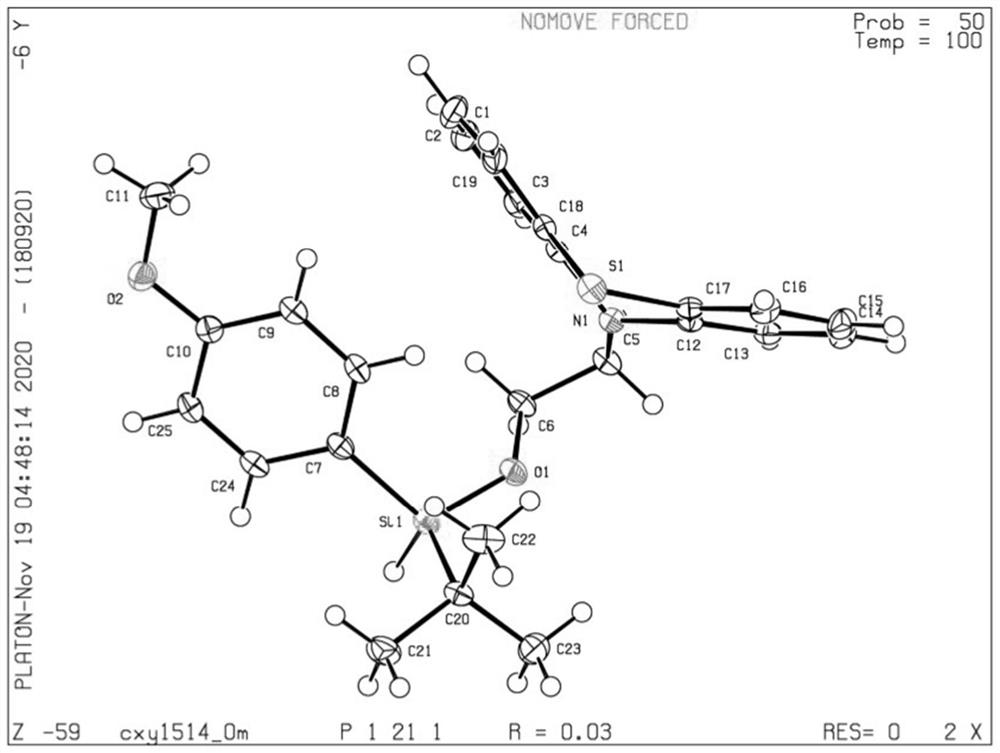
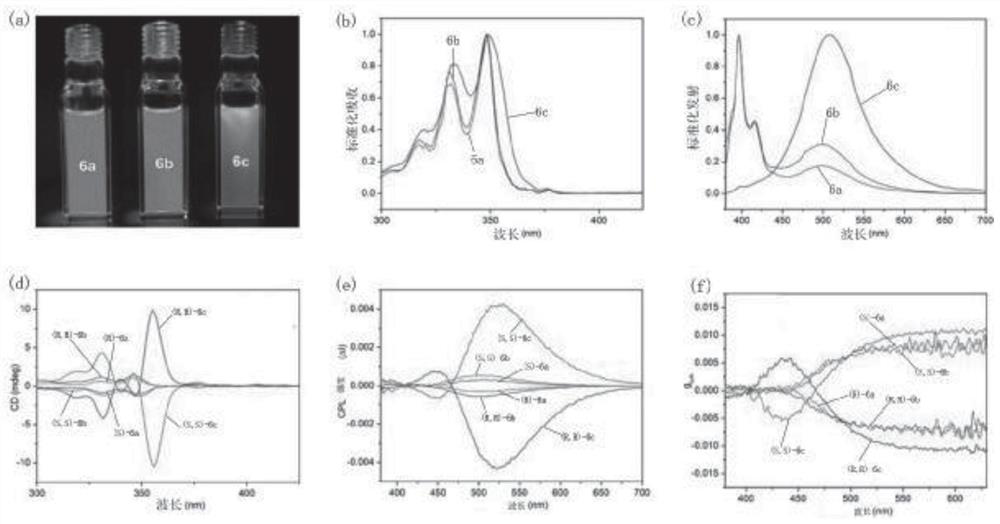
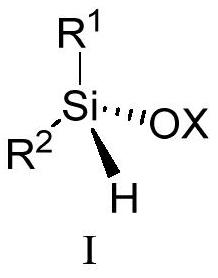
Examples
Embodiment 1
[0087] Synthesis of Dihydrosilane Substrates
[0088]
[0089] Method A: Add tBuLi (1.3M pentane solution, 1.1 equiv) dropwise to aryl bromide (1.0 equiv) in THF (0.5M) at -78°C under the protection of argon, and stir at -78°C 1 h, then add tBuSiCl in 10 mL THF 3 (1.2 equivalents). The mixture was stirred for an additional 1 h at -78 °C, then allowed to warm to room temperature and stirred overnight. The reaction was cooled again to -78 °C and LiAlH was added dropwise 4 (2.5M in THF, 2.0 eq.) and stirred at -78°C for a further 30 min, then the mixture was allowed to warm to room temperature and stirred for 8 h. Finally, the reaction mixture was heated at -78 °C with saturated NH 4 Cl was quenched, and after warming to room temperature, the mixture was filtered and washed with Et 2 Washed with O, the organic layers were combined and washed with MgSO 4 Dry, filter and evaporate under reduced pressure. The crude mixture was purified by column chromatography on silica ge...
Embodiment 2
[0107] Synthesis of silanol and alcohol substrates
[0108]
[0109] Method D: Add nBuLi (2.5M in hexane, 1.1 equiv) dropwise to aryl bromide (1.0 equiv) in THF (0.5M) at -78°C under the protection of argon, and the mixture was cooled at -78°C After stirring for 1 h, chlorodimethylsilane (1.2 equiv) was added and the mixture was stirred at -78 °C for 1 h, then allowed to warm to room temperature and stirred overnight. Finally, the reaction mixture was heated at -78 °C with saturated NH 4 Quenched by Cl, after warming to room temperature, the mixture was filtered and washed with Et 2 Washed with O, the organic layers were combined, washed with MgSO 4 Drying, filtration, and evaporation under reduced pressure afforded the corresponding product, which was used in the next step without further purification.
[0110] Method E: Add KMnO to a solution of silane in THF (0.5M) under air atmosphere 4 (1.5 eq), the mixture was stirred at room temperature for 24 h, filtered through...
Embodiment 3
[0122] Screening of reaction conditions between dihydrosilane and silanol:
[0123] In the presence of Rh catalyst, tert-butyl(4-methoxyphenyl)silane 1a and dimethyl(phenyl)silanol 2a undergo intermolecular dehydrogenation Si-O coupling reaction at room temperature in toluene solvent, Tried several chiral diphosphine ligands, using [Rh(cod)Cl] 2 (1mol%) as the catalyst and Josiphos L1 (2.2mol%) as the chiral ligand, the siloxane product 3a was successfully obtained in 36% yield with good enantiomeric control (ee was 74%), others with different electronic The characteristic Josiphos-type ligands (L2 and L3) are also viable ligands.
[0124] The yield and enantioselectivity were significantly improved (68% yield, 97% ee) by using the Josiphos ligand L4 with the interchanged substituents of the phosphorus atom. Further screening of chiral ligands revealed that BINAP L5 and Segphos L6 were also effective for this reaction.
[0125] With L4 ligand, the common solvents DCE (1,2-d...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More