

Preparation method of sitafloxacin side chain intermediate
A sitafloxacin and side chain technology, applied in the field of biopharmaceuticals, can solve the problems of long preparation time, difficult processing, and only a total yield, and achieve high product yield, high process amplification safety, and small environmental impact. Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

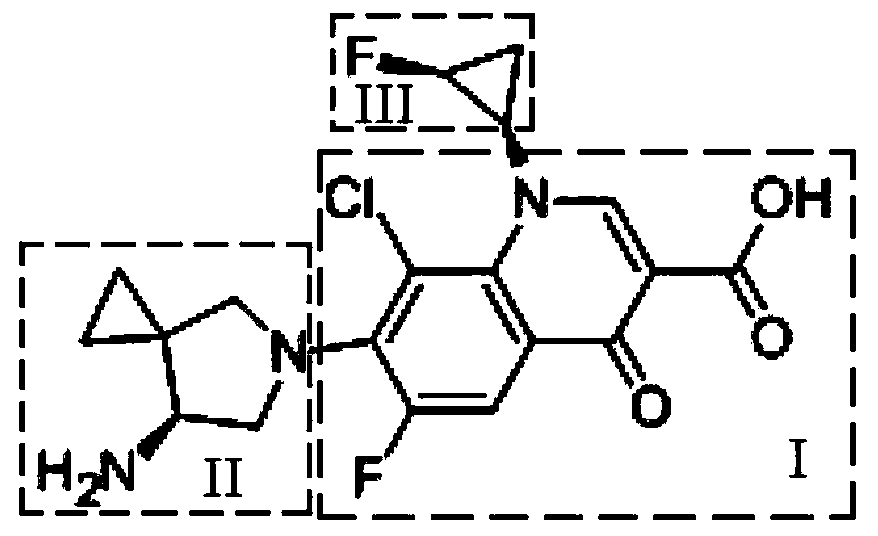
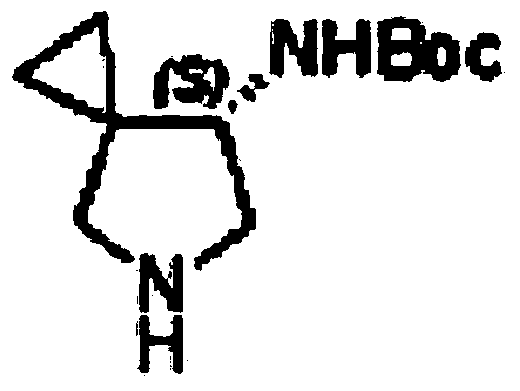
Examples
preparation example Construction
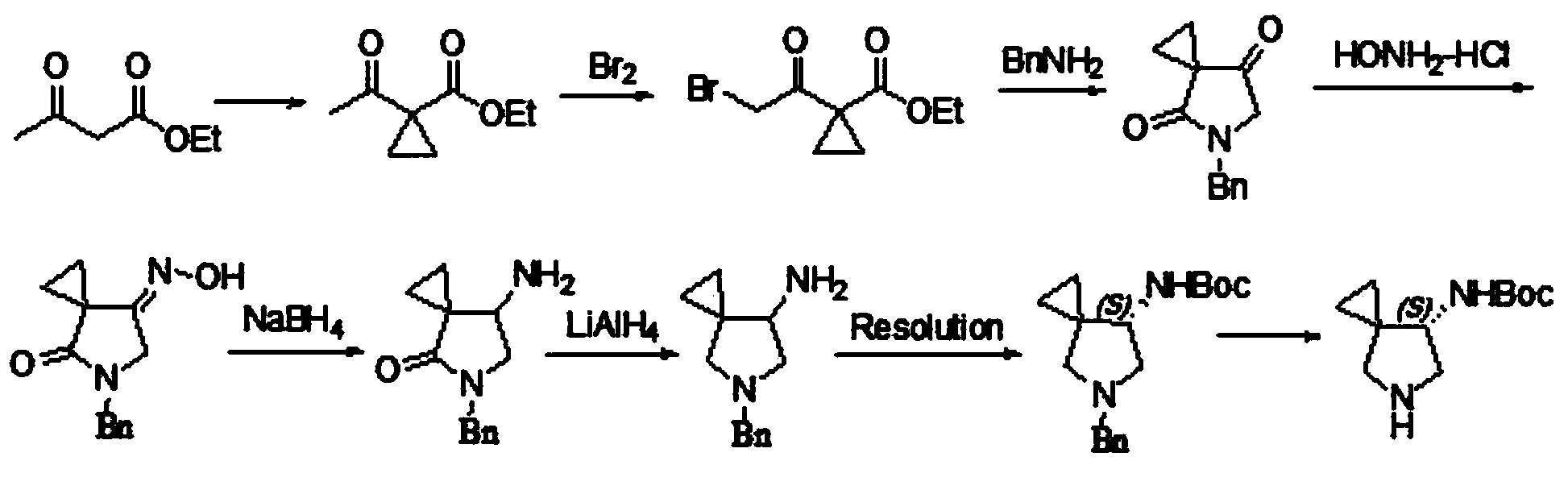
[0033] A method for preparing a sitafloxacin side chain intermediate, comprising the steps of:
[0034] ①Synthesis of a three-membered ring: Dissolve every 1kg of methyl 4-chloroacetoacetate in 5-6L of acetonitrile, add 38-40g of proline, then add 1.25kg of 1,2-dibromoethane, and add 2.3kg of potassium carbonate while stirring -2.5kg, then the reaction solution was stirred at a controlled temperature of 30-36°C for 30-36 hours, filtered, and the filtrate was concentrated to dryness under reduced pressure to obtain a three-membered carbocyclic compound A;
[0035] ②Synthesis of nitrogen heterocycles: Slowly add the obtained A into 5L of ammonia water with a concentration of 25%, stir at room temperature for 2.5-3 hours, then heat up to 38-40° and react for 8-10 hours, then suction filter, wash and dry to obtain Five-membered nitrogen heterocyclic compound B;
[0036] ③Generation of chiral carbon: Dissolve B in 2-2.5L toluene, add R-1-phenylethylamine with 111% of the mass of B...
Embodiment 1
[0042] ①Dissolve 1kg of methyl 4-chloroacetoacetate in 6L of acetonitrile, add 38g of proline and 1.25kg of 1,2-dibromoethane, add a total of 2.3kg of potassium carbonate in 10 times under stirring, and then the reaction solution is heated at 35°C Stir for 36 hours. After filtration, the filtrate was concentrated under reduced pressure to obtain three-membered carbocyclic compound A as a colorless oil.
[0043] ②Continuously add the above A into 5L of 25% ammonia water, stir for 3 hours, then raise the temperature to 40°C, and continue the reaction for 10 hours. Suction filtration, washing the solid with water, and drying to obtain 300 g of five-membered nitrogen-heterocyclic compound B in the form of white solid powder.
[0044] ③Dissolve the above powder B in 2L of toluene, then add 333g of R-1-phenylethylamine, and reflux for 10 hours while stirring. Cooled, concentrated to dryness under reduced pressure to obtain solid chiral compound C.
[0045] ④ Add the above solid C...
Embodiment 2
[0049] ① Dissolve 10kg of methyl 4-chloroacetoacetate in 50L of acetonitrile, add 400g of proline and 12.5kg of 1,2-dibromoethane, add a total of 24kg of potassium carbonate in 9 times under stirring, and then stir the reaction solution at 30°C 36 hours. After filtration, the filtrate was concentrated under reduced pressure to obtain three-membered carbocyclic compound A as a colorless oil.
[0050] ②The above three-membered carbocyclic compound was continuously added to 5L of 25% ammonia water, stirred for 2.5 hours, then heated to 38°C, and continued to react for 8 hours. Suction filtration, washing the solid with water, and drying to obtain 3078 g of five-membered nitrogen heterocyclic compound B in the form of white solid powder.
[0051] ③ Dissolve the above powder B in 25L of toluene, then add 3.4kg of R-1-phenylethylamine, stir and reflux to separate water for 12 hours. Cooled, concentrated to dryness under reduced pressure to obtain solid chiral compound C.
[0052]...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More