

Method for synthesis of favipiravir
A synthetic method, the technology of favipiravir, applied in the field of drug synthesis, can solve the problems of difficult industrial production requirements, low yield, low efficiency, etc., and achieve the effect of simple operation, high yield, and simplified preparation process
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

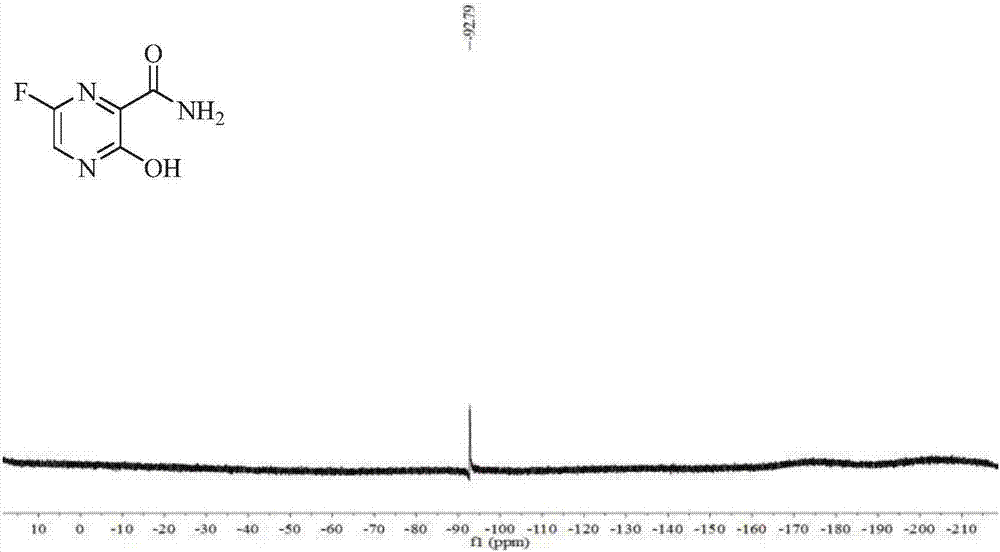
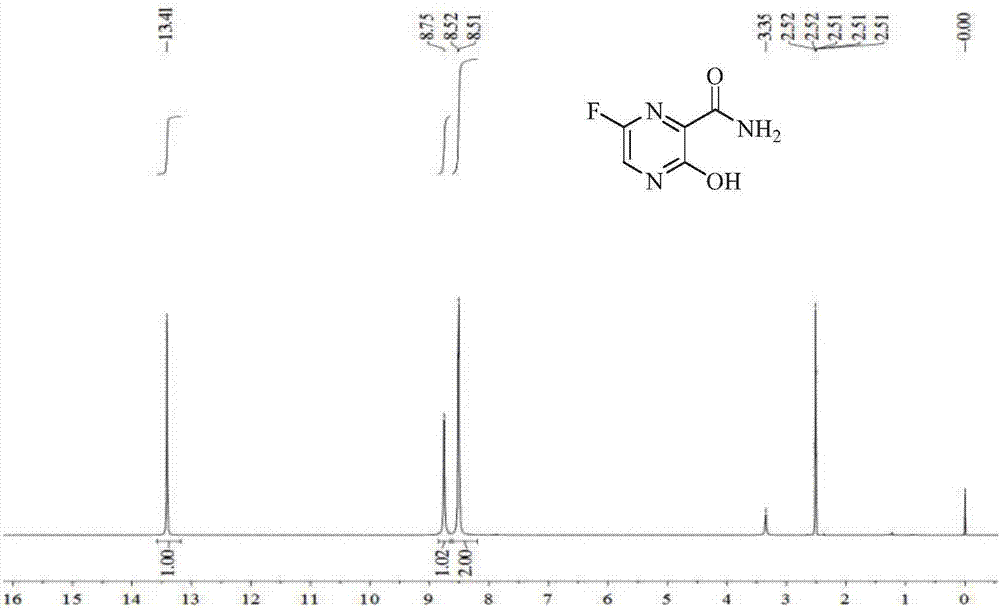
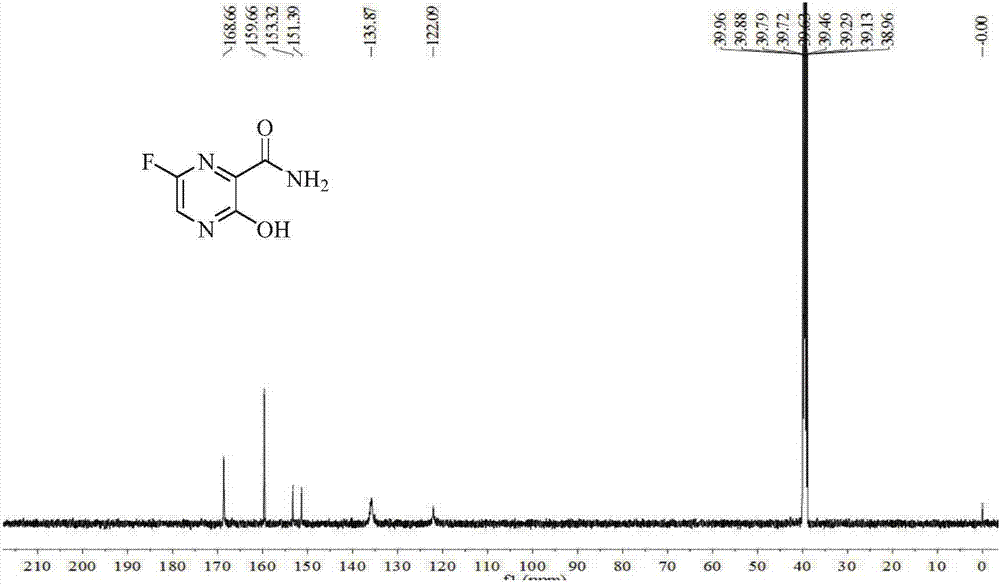
Examples
Embodiment 1
[0138] The first step: preparation of 3-amino-2-ester pyrazine (2)
[0139]
[0140] Add methanol (50ml) to compound 1 (5g, leq), add concentrated sulfuric acid (4eq) under an ice-water bath, stir at room temperature, TLC shows that the reaction is complete, concentrate, adjust the pH to 8 with saturated sodium carbonate, filter with suction, and dry at 50°C for 2h. Obtained 2 as a brown solid (4.18 g, 76%).
[0141] The second step: preparation of methyl 3-amino-6-bromopyrazine-2-carboxylate (3)
[0142]
[0143]Add acetonitrile (276ml) to compound 2 (27.6g, leq), stir at room temperature, add NBS (25.1g, 1.01eq) in batches, stir overnight at room temperature, TLC shows that after the reaction is complete (20-30h), add water (300ml), use Na 2 CO 3 The solution was adjusted to pH=7, extracted with ethyl acetate (3×50ml), the organic phases were combined, dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure to obtain 3...
Embodiment 2
[0163] Compared with Example 1, the difference is only in:
[0164] The fifth step: preparation of 3,6-dichloropyrazine-2-carbonitrile (6)
[0165] Compound 5 (2g, 1eq) was dissolved in chlorobenzene (10ml), POCl was slowly added dropwise 3 (5.6g, 4eq), heated to 70°C to form a homogeneous solution, lowered to room temperature, added DIEA (3.57g, 3eq) dropwise, stirred for 1h at 60°C, 1h at 80°C, and 4h at 100°C. Afterwards, it was poured into ice water (110ml) and stirred vigorously to react for 2h, suction filtered, and 20 times of petroleum ether was used for the filter cake (the weight ratio of the obtained crude product to petroleum ether was 1:10-30) to obtain a brown solid 6 in a yield of 60 %.
[0166] The remaining steps are the same as in Example 1.
Embodiment 3
[0168] Compared with embodiment 1, the difference only lies in:
[0169] The crude product of 3-amino-6-bromopyrazine-2-carboxylic acid methyl ester obtained in the second step was added 30 times of dichloromethane (the weight ratio of the obtained crude product to dichloromethane was 1:25~50) and refluxed for 0.5h Afterwards, suction filtration, the dichloromethane was evaporated from the mother liquor, and then recrystallized with 20 times of methanol (the weight ratio of rotary steamed product and methanol was 1:5-30) to obtain light yellow solid 3 with a yield of 76%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More