

Synthesis method of oseltamivir
A synthesis method and technology of oseltamivir are applied in the field of preparation of drug oseltamivir, which can solve the problems of many reaction steps, high cost, low total yield and the like, and achieve the effects of easy recovery and short route.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
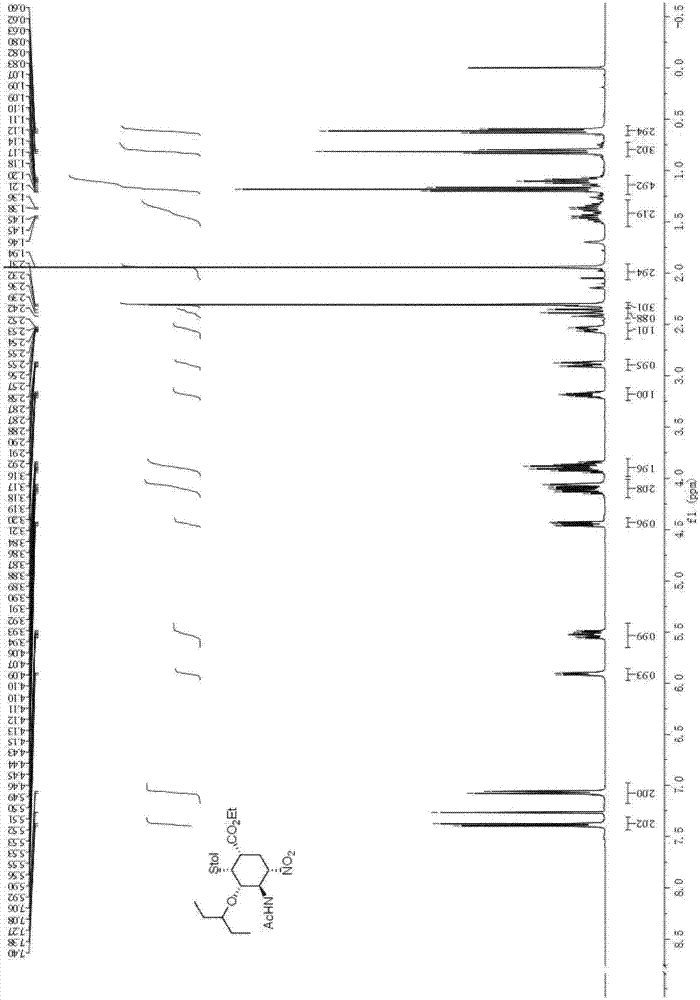
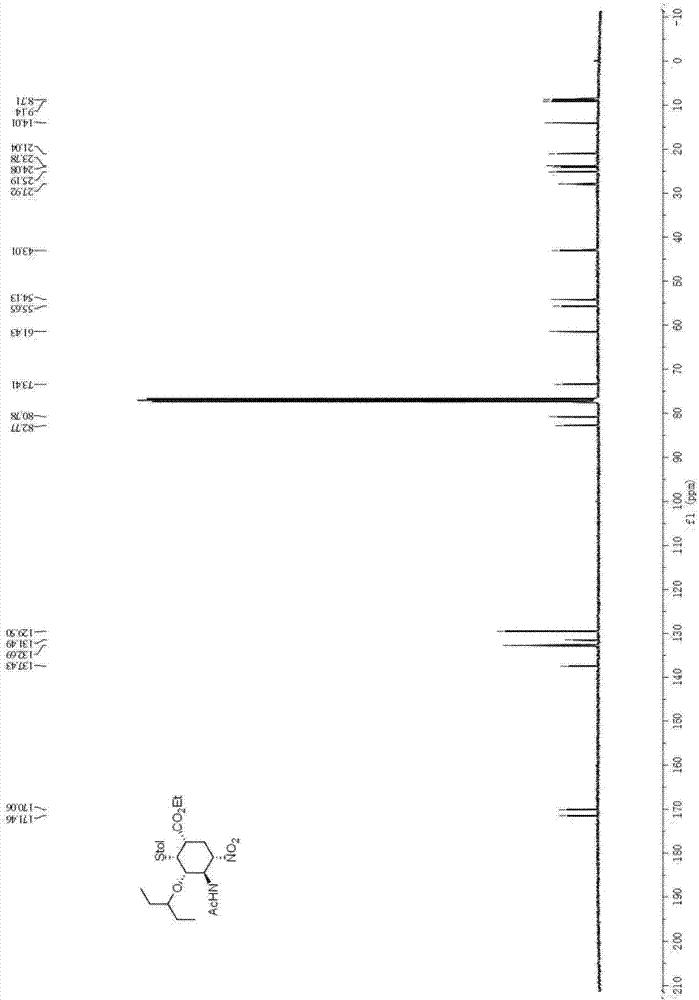
[0040] (1) Synthesis of Intermediate C:
[0041]
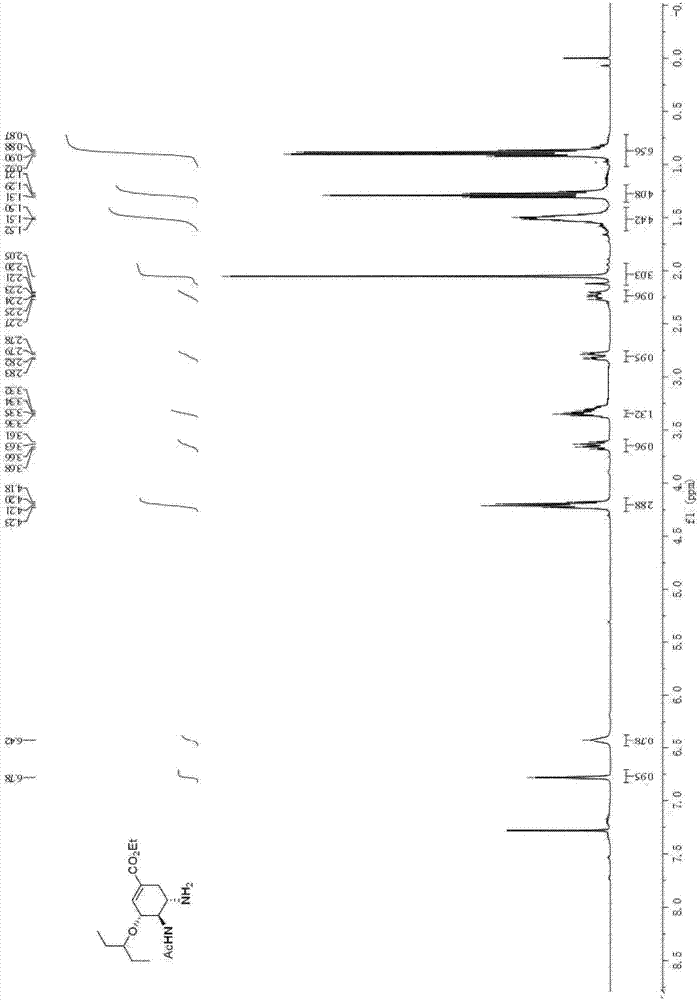
[0042] Add dichloromethane (6ml) in reaction bottle, then add acetamido nitroalkene (260mg, 2 mmol), 3-pentyloxyacetaldehyde (520mg, 4mmol) and chloroacetic acid (76mg, 0.8mmol), finally N,N-Dimethylbenzylaminoprolinol trimethylsilyl ether catalyst (88mg, 0.2mmol) was added, and the resulting mixture was stirred at room temperature for 2 hours. The reaction system was cooled to 0°C, and 2-diethoxyethylphosphonoacrylate (0.95g, 4mmol) and cesium carbonate (1.60g, 5mmol) were added thereto, and the reaction was stirred at 0°C for 3 hours, and then decompressed Remove the solvent, then add absolute ethanol (8mL), stir at room temperature for 15 minutes, then add p-cresylthiophenol (1.0g, 8mmol) at -15°C, stir the reaction system at room temperature for 48 hours, then wash it with cold 2 equivalents of hydrochloric acid Quenched, the aqueous phase was extracted three times with dichloromethane (10mL), the combined organic phas...
Embodiment 2
[0049] (1) Synthesis of Intermediate C:
[0050] Add dichloromethane (6ml) in the reaction flask, then add acetamidonitroolefin (260mg, 2 mmol), 3-pentyloxyacetaldehyde (520mg, 4mmol) and o-nitrobenzoic acid (134mg, 0.8mmol ), and finally N, N-dimethylbenzylamine prolinol trimethylsilyl ether catalyst (88mg, 0.2mmol) was added, and the resulting mixture was stirred at room temperature for 3 hours. The reaction system was cooled to 0°C, and 2-diethoxyethylphosphonoacrylate (0.95g, 4mmol) and cesium carbonate (1.60g, 5mmol) were added thereto, and the reaction was stirred at 0°C for 3 hours, and then decompressed Remove the solvent, then add absolute ethanol (8mL), stir at room temperature for 15 minutes, then add p-cresylthiophenol (1.0g, 8mmol) at -15°C, stir the reaction system at room temperature for 48 hours, then wash it with cold 2 equivalents of hydrochloric acid Quenching, the aqueous phase was extracted three times with dichloromethane (10mL), the combined organic pha...
Embodiment 3
[0054] (1) Synthesis of Intermediate C:
[0055] Add chloroform (6ml) in reaction bottle, then add acetamido nitroalkene (260mg, 2 mmol), 3-pentyloxyacetaldehyde (520mg, 4mmol) and benzoic acid (98mg, 0.8mmol), finally N,N-Dimethylbenzylaminoprolinol trimethylsilyl ether catalyst (88mg, 0.2mmol) was added, and the resulting mixture was stirred at room temperature for 2 hours. The reaction system was cooled to 0°C, and 2-diethoxyethyl phosphoacrylate (0.95g, 4mmol) and cesium carbonate (1.60g, 5mmol) were added thereto, and the reaction was stirred at 0°C for 3 hours, and then depressurized Remove the solvent, then add absolute ethanol (8mL), stir at room temperature for 15 minutes, then add p-cresylthiophenol (1.0g, 8mmol) at -15°C, stir the reaction system at room temperature for 48 hours, then wash it with cold 2 equivalents of hydrochloric acid Quenched, the aqueous phase was extracted three times with dichloromethane (10mL), the combined organic phase was washed with 15mL...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More