

High-solubility photochromic compound and preparation method thereof
A photochromic, high-solubility technology, applied in chemical instruments and methods, color-changing fluorescent materials, organic chemistry, etc., can solve the problems of lack of flexible substituents, affect product quality, poor compatibility, etc., and achieve easy processing Film-forming, no background color, excellent solubility effects
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

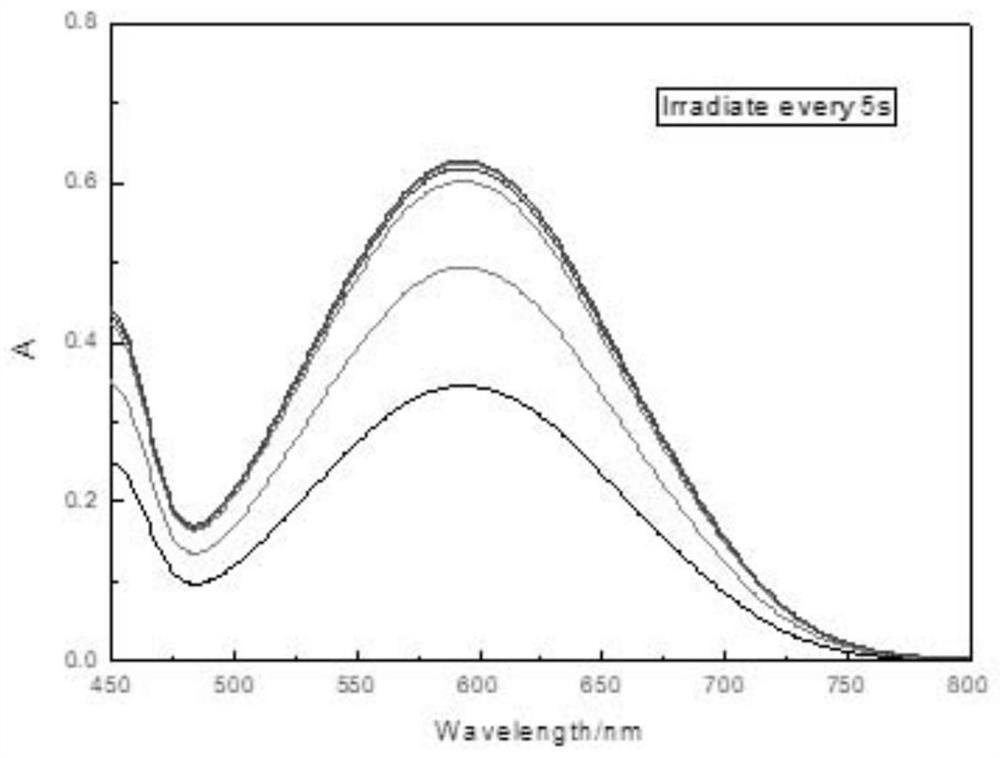
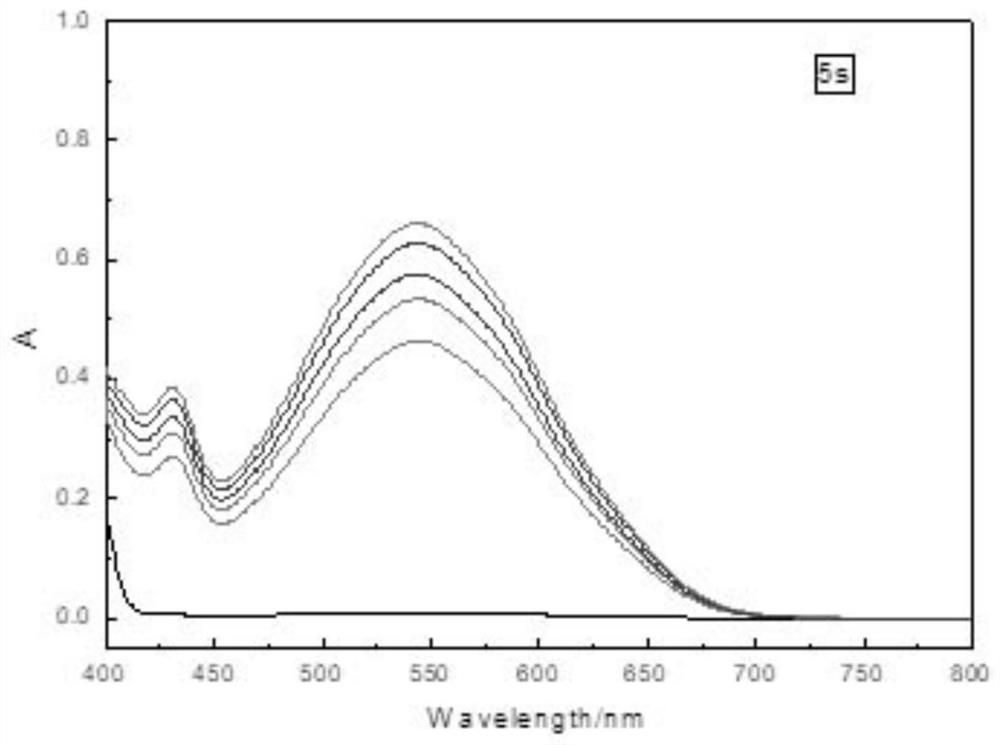
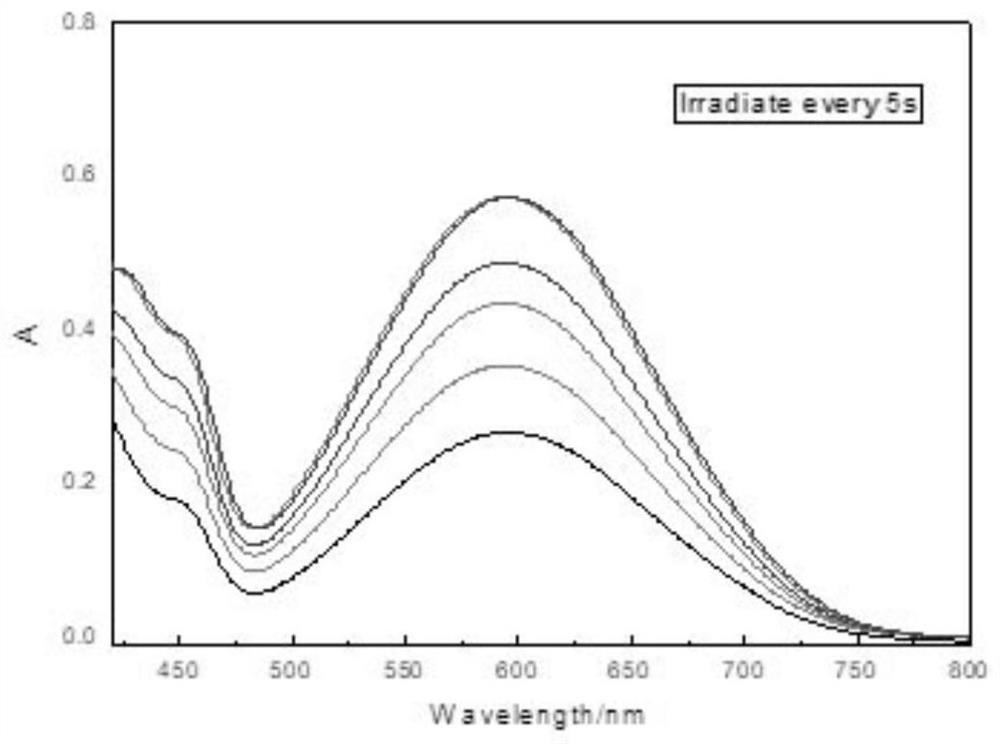
Examples
Embodiment 1
[0062] The synthesis of embodiment 1 photochromic compound Ia
[0063] Step 1: Synthesis of Compound C1
[0064]
[0065] To a 100 mL Shrek tube at 0°C, sodium hydride (0.2 g, 64 mmol) was added, followed by 30 mL of anhydrous tetrahydrofuran. 4,4'-Dihydroxybenzophenone A1 (2.1 g, 10 mmol) was dissolved in 30 ml of anhydrous DMF, and slowly added into the reaction vial with a syringe, and stirred for 1 h. B1 (1.5 g, 64 mmol) was added to the reaction system, heated to 60° C. for 72 h. Cool to room temperature, add saturated NH to the reaction system 4 Cl solution was extracted three times with ethyl acetate (3×50ml), and the organic phases were combined and washed once with brine (50mL) and water (50mL) respectively. Dry over anhydrous sodium sulfate and filter. The solvent was removed by a rotary evaporator to obtain a crude product, which was separated and purified by silica gel column chromatography (eluent: dichloromethane / methanol = 40:1) to obtain a yellow oil C1 ...
Embodiment 2
[0077] The synthesis of embodiment 2 photochromic compounds Ib
[0078] Step 1: Synthesis of Benzophenone Derivative C2
[0079]
[0080] Weigh sodium hydride powder (1.9 g, 80 mmol), add it into a 250 mL Shrek bottle, and add 50 mL of anhydrous tetrahydrofuran under nitrogen protection. Dissolve A (25.71g, 25mmol) in 25mL of anhydrous N,N-dimethylformamide, and drop it into the reaction flask after fully dissolved. Ice bath, stirring for one hour, B1 (13.7g, 50mmol) was added to the reaction flask. Heated to 60°C and reacted for 72 hours. Cool to room temperature, add saturated NH to the reaction system 4 Cl solution was extracted three times with ethyl acetate (3×50ml), and the organic phases were combined and washed once with brine (50mL) and water (50mL) respectively. Dry over anhydrous sodium sulfate and filter. The solvent was removed by a rotary evaporator to obtain a crude product, which was separated and purified by silica gel column chromatography (eluent: di...
Embodiment 3
[0092] Embodiment 3: the synthesis of photochromic compound Ic
[0093] D3 synthetic reference embodiment 1 and 2 steps, no longer repeat
[0094]
[0095] Add D3 (320mg, 1mmol), E2 (300mg, 1mmol) and 20mL toluene to a 100mL single-necked bottle, and stir. Add 2 drops of dodecylbenzenesulfonic acid with a rubber dropper and heat to 40°C for 3h. Cool to room temperature, extract three times with dichloromethane (3×20ml), combine the organic phases, and wash once with saturated brine (20ml) and distilled water (20ml) respectively. Dry over anhydrous magnesium sulfate, filter, and remove the solvent with a rotary evaporator. The residue is separated and purified by silica gel column chromatography (eluent: dichloromethane / methanol = 100:1) to obtain a light yellow solid Ic with a yield of 79%.
[0096] H NMR spectrum of Ic: 1 H NMR (400MHz, CDCl 3 )δ(ppm)8.52(d,J=8.0Hz,1H),8.07(d,J=8.0Hz,1H),7.75(d,J=8.0Hz,1H),7.54–7.42(m,4H), 7.30–7.24(m,2H),7.05–7.02(m,1H),6.95–6.90(m,1...
PUM
| Property | Measurement | Unit |
|---|---|---|
| melting point | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More