

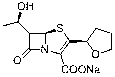
Preparation method of faropenem
A technology of faropenem and tert-butyldimethylsiloxyethyl, which is applied in the field of pharmaceutical synthesis, can solve the problems of unfavorable large-scale industrial production, excessive heavy metal residues and high cost, and achieves shortened production cycle, high reaction yield and cost. low effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

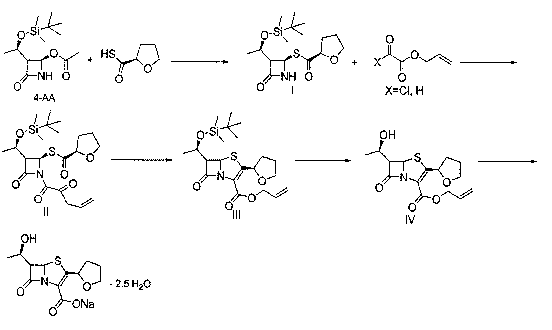
Examples
Embodiment 1
[0027] Preparation of tert-butoxyoxalyl chloride
[0028] Mix tert-butanol (89g, 1.2mol) and an appropriate amount of triethylamine (139g, 1.25mol) and dissolve in 300mL of dichloromethane, slowly add dropwise to oxalyl chloride diluted with 200mL of dichloromethane at -10°C ( 152g, 1.2mol), the dropwise addition temperature was controlled within the range of -10~0°C, and the dropwise addition was completed within 2 hours. Maintain the reaction at -10~0°C for 1 hour, stop the reaction, and let the temperature rise to room temperature naturally. The reaction solution was distilled under reduced pressure to obtain 164 g of tert-butoxyoxalyl chloride, which was detected by gas chromatography with a purity of 95%.
Embodiment 2
[0030] (3R,4R)-3-[(R)-1-tert-butyldimethylsiloxyethyl]-4-[(R)-tetrahydrofuran-2-formylthio]azetidin-2- Preparation of Ketone (Intermediate 1)
[0031] Dissolve R-(+)-thiotetrahydrofuran-2-carboxylic acid (158g, 1.2mol) in 100mL of dichloromethane, dilute triethylamine (139g, 1.25mol) with 50mL of dichloromethane and add dropwise to In the above solution, the pH value was 9 after dropping, and the stirring was continued for 0.5 hour, and it was set aside.
[0032] (3R, 4R)-3-[(R)-1-tert-butyldimethylsiloxyethyl]-4-[(R)-acetoxy]azetidin-2-one (287g, 1mol) was dissolved in 500mL of dichloromethane, zinc chloride (142g, 1.045mol) was added at room temperature, after stirring for 15 minutes, triethylamine of R-(+)-thiotetrahydrofuran-2-carboxylic acid prepared above was added For salt, the temperature during the dropwise addition should not be higher than 30°C. After dropping, react at room temperature for 8 hours. Stop the reaction, pour the reaction solution into 1000mL 3% so...
Embodiment 3
[0034] (3R,4R)-1-tert-butoxyoxalyl-3-[(R)-1-tert-butyldimethylsiloxyethyl]-4-[(R)-tetrahydrofuran-2-formyl Preparation of Thio]azetidin-2-one (Intermediate 2)
[0035] Intermediate 1 (323g, 0.9mol) was dissolved in 500mL of dichloromethane, cooled to -10°C, tert-butoxyoxalyl chloride (164g, 0.95mol) prepared in Example 1 was added dropwise, and the dropwise was completed. Triethylamine (145mL, 1.04mol) was diluted with 200mL of dichloromethane and added dropwise to the above reaction solution, the rate of addition was controlled so that the internal temperature was not higher than -5°C. After dropping, keep the reaction for 1.5 hours, stop the reaction, wash with water, 5% sodium bicarbonate solution, and saturated brine, dry over anhydrous sodium sulfate, and concentrate to obtain a light yellow oil, which is recrystallized from dichloromethane / petroleum ether , to obtain 414.5 g of pale yellow needle-like crystals. 1 H-NMR (CDCl3, 500MHz) δ: 0.41 (s, 6H), 1.01 (s, 9H), 1.3...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More