

Method for preparing Roxadustat intermediate
A technology for intermediates and compounds, which is applied in the preparation of imino compounds, organic chemistry, etc., can solve the problems of difficult quality purification of the final product API, cumbersome preparation process, and high preparation cost, and achieves easy availability of raw materials and high reaction yield. , the effect of fewer steps
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

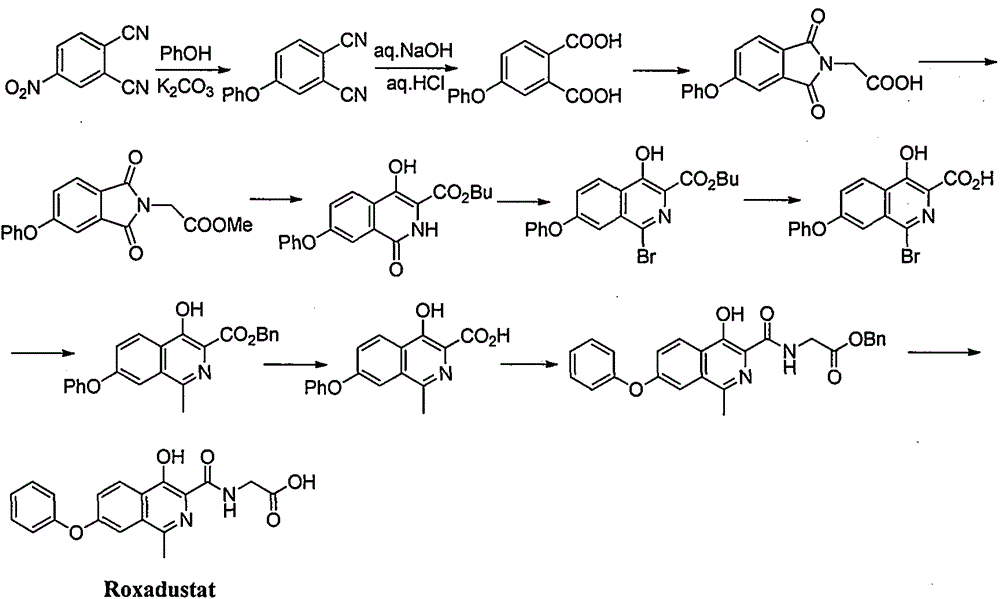
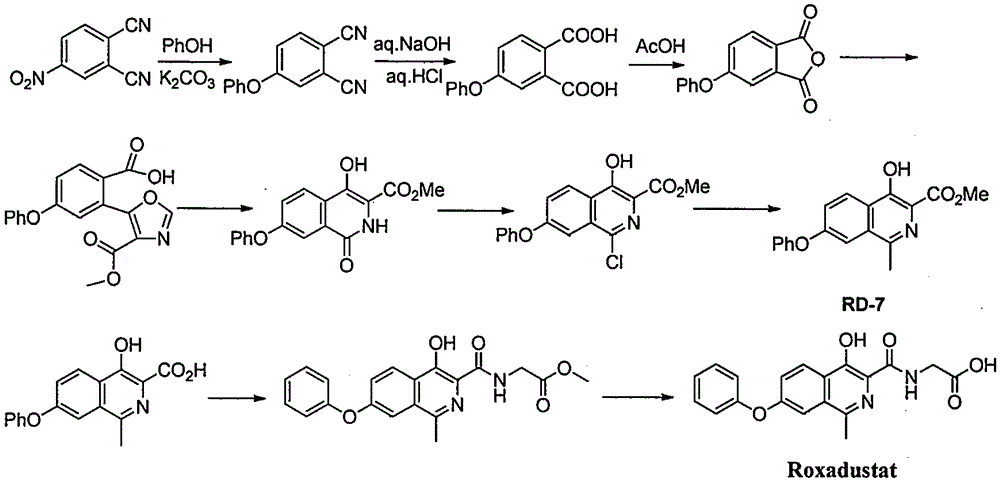
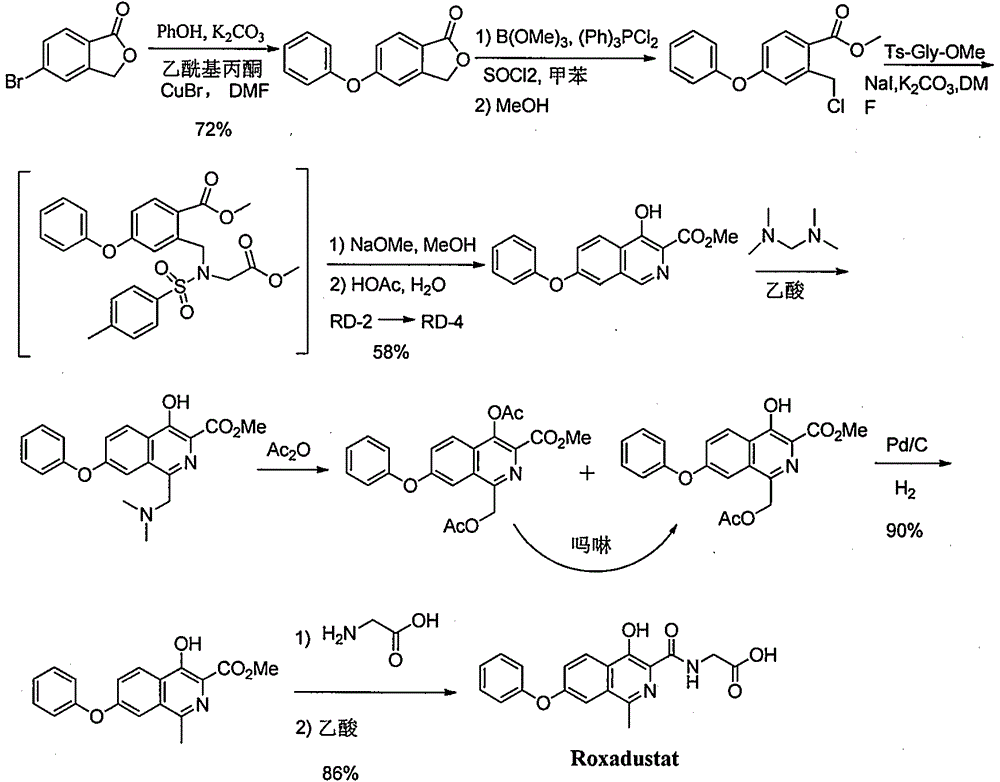
Examples
Embodiment 1
[0035] Preparation of 1-(3-phenoxyphenyl)ethylamine (I)
[0036]
[0037] In a 2L reaction flask, add phenol (188g, 2mol), 3-bromoacetophenone (1,199g, 1mol), cuprous bromide (21.7g, 0.5mol), acetylacetone (10g, 0.1mol), Potassium carbonate (276 g, 2 mol) and N,N-dimethylformamide (1 L). Heated to an internal temperature of 120°C for 7 h, cooled to room temperature, poured the reaction solution into 2N cold aqueous hydrochloric acid (1.5 L), and stirred for 30 min. After filtering, the filter cake was washed with water (0.5 L), dried in vacuum (60° C.) for 5 h, and the compound of formula 2 (188.7 g, yield 89%) was obtained. MS m / z213[M+H] + .
[0038] In the 2L reaction flask, add formula 2 compound (188.7g, 0.89mol), hydroxylamine hydrochloride (74.2g, 1.07mol), sodium hydroxide (71.2g, 1.78mol) and absolute ethanol (0.8L) successively, heat to 60 ℃ reaction 3h, the reaction is complete. After cooling down to room temperature, sodium borohydride (67.6 g, 1.78 mol) wa...
Embodiment 2
[0040] Preparation of Dimethyl 2-(1-(3-phenoxyphenyl)ethylimino)malonate(III)
[0041]
[0042] In the 2L reaction flask, add 1-(3-phenoxyphenyl) ethylamine (1, 175g, 0.82mol), dimethyl ketomalonate (120g, 0.82mol), p-toluenesulfonic acid (7g) successively , 41 mmol) and toluene (1 L). Heated to reflux for 8 hours, cooled to room temperature, added water (0.5L) for extraction, and concentrated the organic layer to dryness under reduced pressure to obtain a crude product, which was recrystallized by adding absolute ethanol (0.5L) to obtain 2-(1-( Dimethyl 3-phenoxyphenyl)ethylimino)malonate (III, 274.5 g, yield 98%). MS m / z 342[M+H] + . 1 H NMR (400Hz, DMSO-d 6 )δ1.25 (d, J=3.2Hz, 3H), 2.95 (m, 1H), 3.71 (s, 6H), 6.98-7.03 (m, 3H), 7.15-7.19 (m, 4H), 7.38-7.41 (m, 2H).
Embodiment 3
[0044] Preparation of Diethyl 2-(1-(3-phenoxyphenyl)ethylimino)malonate(III)
[0045]
[0046] In the 2L reaction flask, add 1-(3-phenoxyphenyl) ethylamine (1, 175g, 0.82mol), diethyl ketomalonate (143g, 0.82mol), methanesulfonic acid (4g) successively , 41 mmol) and xylene (1 L). Heat to 130°C for 6 hours, cool to room temperature, add water (0.5L) for extraction, and concentrate the organic layer to dryness under reduced pressure to obtain a crude product, which is recrystallized by adding absolute ethanol (0.5L) to obtain 2-(1- Diethyl (3-phenoxyphenyl)ethylimino)malonate (III, 291 g, yield 96%). MS m / z 370[M+H] + . 1 H NMR (400Hz, DMSO-d 6 )δ1.21(d, J=3.2Hz, 3H), 1.29(m, 6H), 2.93(m, 1H), 4.22(m, 4H), 6.97-7.01(m, 3H), 7.14-7.17(m , 4H), 7.39-7.41 (m, 2H).
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More