

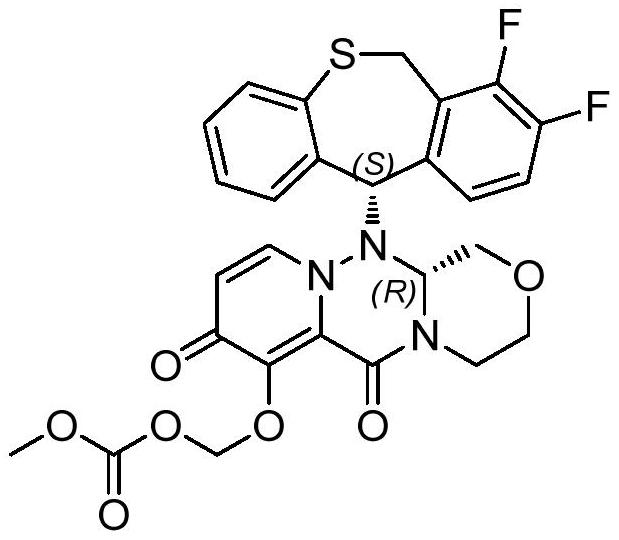
A kind of synthetic method of anti-influenza drug
A synthetic method and suitable technology, applied in the field of medicine and chemical industry, can solve the problems of long steps, high process cost and low overall yield
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
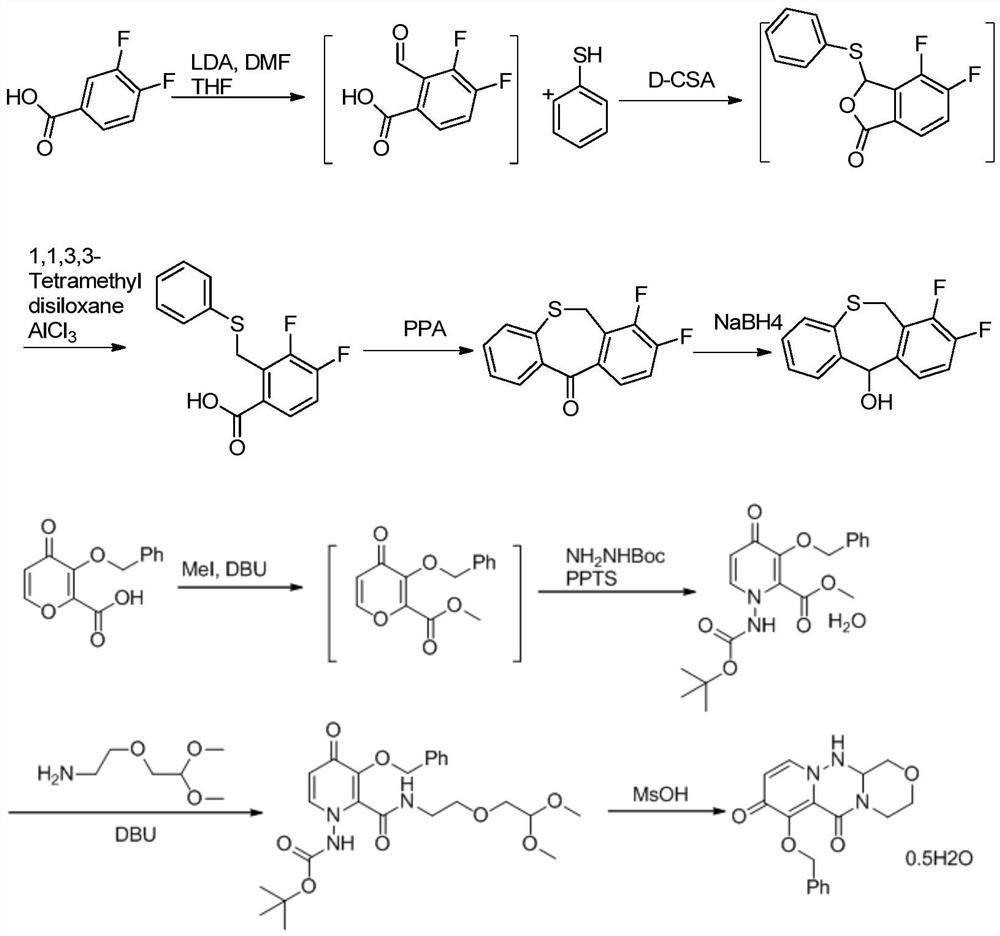
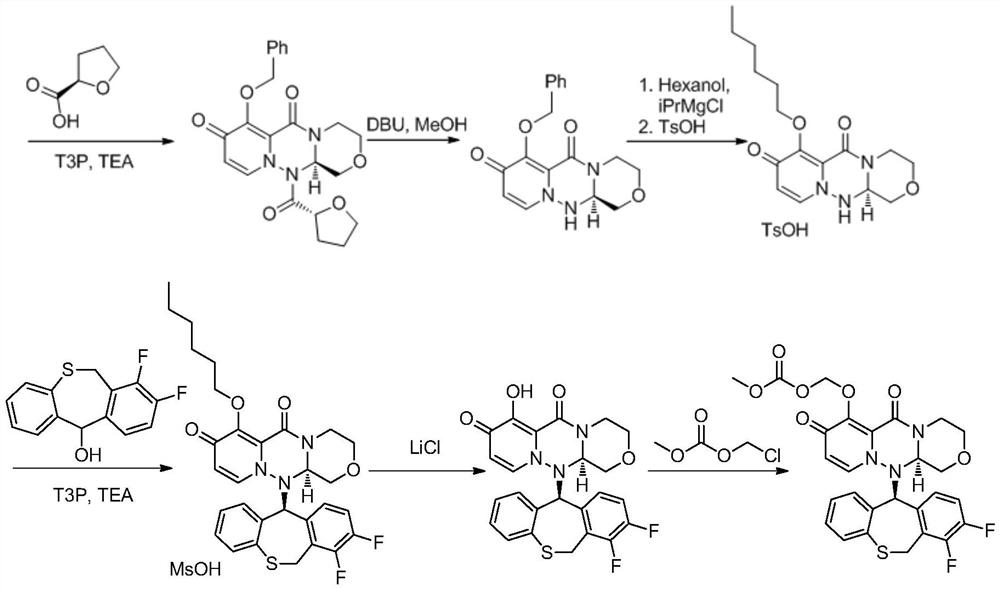
Embodiment 1
[0038]
[0039] Add thiosalicylic acid (15.42g, 100mmol), 1-(chloromethyl)-2,3-difluorobenzene 2 (17.07g, 105mmol) and N,N-dimethylformamide (77mL ), add potassium carbonate (20.73g, 150 mmol), stir well and then heat to 45-55°C for 10-16 hours. After the reaction was completed, water (154 mL) was added, and a large amount of solid precipitated out, which was filtered and dried to obtain compound 3 (26.06 g, yield 93%).
[0040]Here the solvent N,N-dimethylformamide can be replaced by dichloromethane, acetone, acetonitrile, tetrahydrofuran, 2-methyltetrahydrofuran, N,N-dimethylformamide, N-methylpyrrolidone or toluene; Potassium can be replaced by potassium bicarbonate, sodium bicarbonate, sodium carbonate, cesium carbonate inorganic base or triethylamine, diisopropylethylamine, pyridine, DMAP, DBU or DABCO.
Embodiment 2
[0042]
[0043] Add polyphosphoric acid (84mL) into the three-necked flask, heat up to 75-85°C and stir, add compound formula 3 (28.03g, 100mmol), after the addition, heat up to 100-110°C for 3-4 hours, and cool to 75°C after the reaction is complete. Slowly add water (280ml) dropwise at ~85°C to quench the reaction, cool to room temperature, add dichloromethane (140mL) and stir for 20 minutes, then separate the organic phase, extract the aqueous phase with dichloromethane (70mL) twice, and combine the organic phases Wash once with saturated sodium bicarbonate (140mL) solution, once with saturated brine (70mL), dry over sodium sulfate, concentrate, beat with petroleum ether, filter, and dry to obtain compound 4 (24.13g, yield 92%).
[0044] In this reaction step, solvents such as dichloromethane, 1,2-dichloroethane, tetrahydrofuran, 2-methyltetrahydrofuran or 1,4-dioxane can also be added as the reaction solvent.
Embodiment 3
[0046]
[0047] Add compound formula 3 (28.03g, 100mmol) and dichloromethane (140mL) into the three-necked flask, stir and dissolve, add thionyl chloride (17.85g, 150mmol) dropwise, heat and reflux for 4 to 6 hours, spin off after the reaction Most of the dichloromethane, then add dichloromethane (56mL) and swirl once, then add dichloromethane (140mL) and stir to dissolve, cool to 0-5°C, add aluminum trichloride (26.67g, 200mmol) in batches, After adding, it was warmed up to room temperature and reacted for 3 to 5 hours. After the reaction, water (280ml) was added dropwise to quench the reaction. The organic phase was separated, and the aqueous phase was extracted twice with dichloromethane (70mL). The organic phase was combined with saturated sodium bicarbonate ( 140mL) solution, washed once with saturated brine (70mL), dried over sodium sulfate, concentrated and then slurried with petroleum ether, filtered, and dried to obtain compound formula 4 (24.13g, yield 87%).
[00...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More