

Preparation method of baricitinib
A baricitinib and intermediate technology, applied in the field of drug synthesis, can solve problems such as product loss and shorten reaction steps, and achieve the effects of low cost, easy availability of raw materials, and convenient operation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

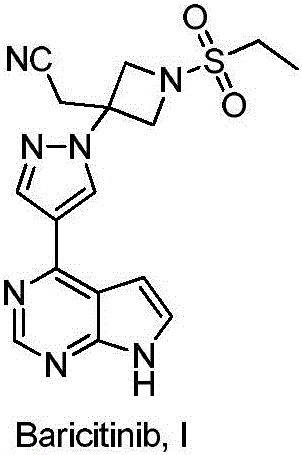
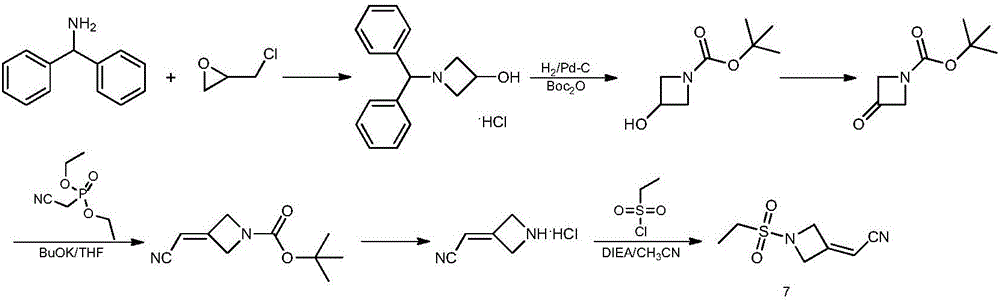
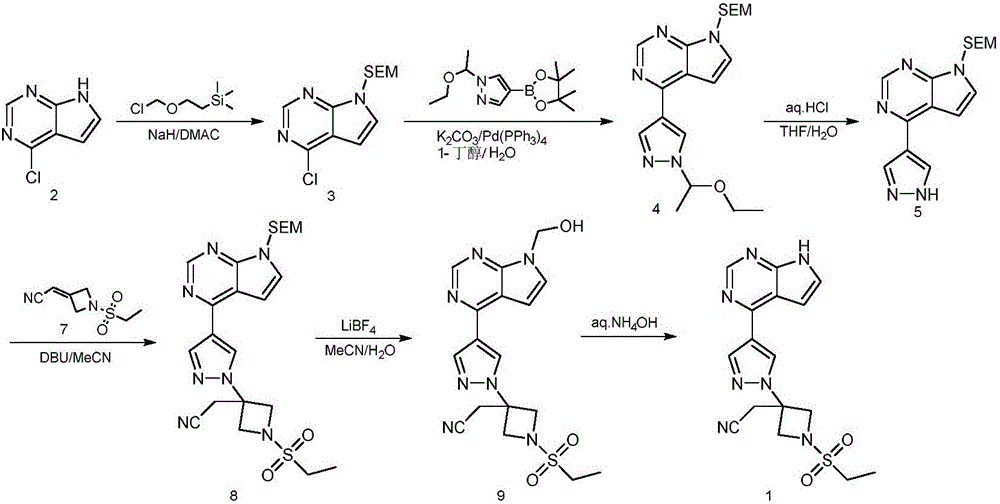
Examples
Embodiment 1
[0034] Preparation of 4-chloro-7-(phenylsulfonyl)-7H-pyrrolo[2,3-d]pyrimidine (III)
[0035] 4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (II, 15.00g, 0.098mol) was dissolved in 400mL THF, then potassium tert-butoxide (13.70g, 0.122mol) was added, and the reaction mixture was kept at room temperature Stir for 20 min. When the reaction exotherms, use an ice-water bath to assist cooling, add benzenesulfonyl chloride (15.6 mL, 0.122 mol) dropwise to the mixed solution, and stir for 3 hours after dropping. The reaction was stopped. After removing part of the solvent under reduced pressure, the reaction solution was slowly poured into 600mL ice-water mixture and stirred for 30 min. A large amount of solid was washed out. After filtration, it was vacuum dried to obtain a white solid, namely 4-chloro-7-(phenylsulfonyl)- 7H-pyrrolo[2,3-d]pyrimidine (25.64 g, yield 89.1%). MS m / z 294[M+1] + ; 1 H-NMR(400M,CDCl 3 ) δ: 8.78 (s, 1H), 8.22 (d, 2H), 7.65 (t, 1H), 7.57-7.55 (t, 1H), 6.73-6.72 (d, 1H) ...
Embodiment 2
[0037] Preparation of 7-(phenylsulfonyl)-4-(1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidine (V)
[0038] At room temperature, 4-chloro-7-(phenylsulfonyl)-7H-pyrrolo[2,3-d]pyrimidine (III, 10.00g, 0.034mol), water (50mL) and potassium carbonate (14.12g, 0.102mol) was added to a 500mL reaction flask, and then 4-pyrazoleboronic acid pinacol ester (IV, 7.93g, 0.041mol), DMF (100mL) and Pd (PPh 3 ) 4 (0.59 g, 0.511 mmol). The reaction mixture was heated to 80-85°C, and then kept warm and stirred for 4 hours. The reaction was monitored by TLC. After the reaction was complete, add 500 mL of ice-water mixture to the reaction flask and stir for 20 min. The solid was precipitated. After filtration, the crude product was dried. Then the crude product was dissolved by heating with 50 mL of isopropanol. Allow to cool to precipitate a solid, filter, and dry in vacuo to obtain a white solid, namely 7-(phenylsulfonyl)-4-(1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidine (9.40g, The yield is 85.0%). M...
Embodiment 3
[0040] 3-(cyanomethyl)-3-(4-(7-benzenesulfonyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1Hpyrazol-1-yl)aza Preparation of tert-butyl cyclobutane-1-carboxylate (VII)
[0041] At room temperature, add DMF (100mL), 7-(benzenesulfonyl)-4-(1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidine (V , 9.40g, 0.029mol) and tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (VI, 6.20g, 0.032mol), stir and mix well, then add DBU dropwise to the reaction system (0.221g, 1.452mmol), continue to stir at room temperature for 14 hours. When the reaction was complete, the reaction mixture was quenched with water (150 mL) and acetonitrile (100 mL), the resulting mixture was stirred at room temperature for 30 min, filtered, and the solid was collected and washed with acetonitrile-water mixture (2:3v / v, 20mL×2). Dry under vacuum at 40-45℃ to obtain white solid, namely 3-(cyanomethyl)-3-(4-(7-benzenesulfonyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl) -1H pyrazol-1-yl)azetidine-1-carboxylic acid tert-butyl ester (13.86 g,...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More