

Targeted traceless release drug conjugate as well as preparation method and application thereof
A technology of conjugates and drugs, which can be used in drug combinations, pharmaceutical formulations, anti-tumor drugs, etc., can solve the problems of low release efficiency of enzyme cleavage response complexes and the need to improve tumor-inhibitory activity, and achieve novel linker structure and reduce toxicity. , enhance the effect of treatment
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1R
[0059] Example 1 Synthesis of RGD-SS-CA (2)
[0060]
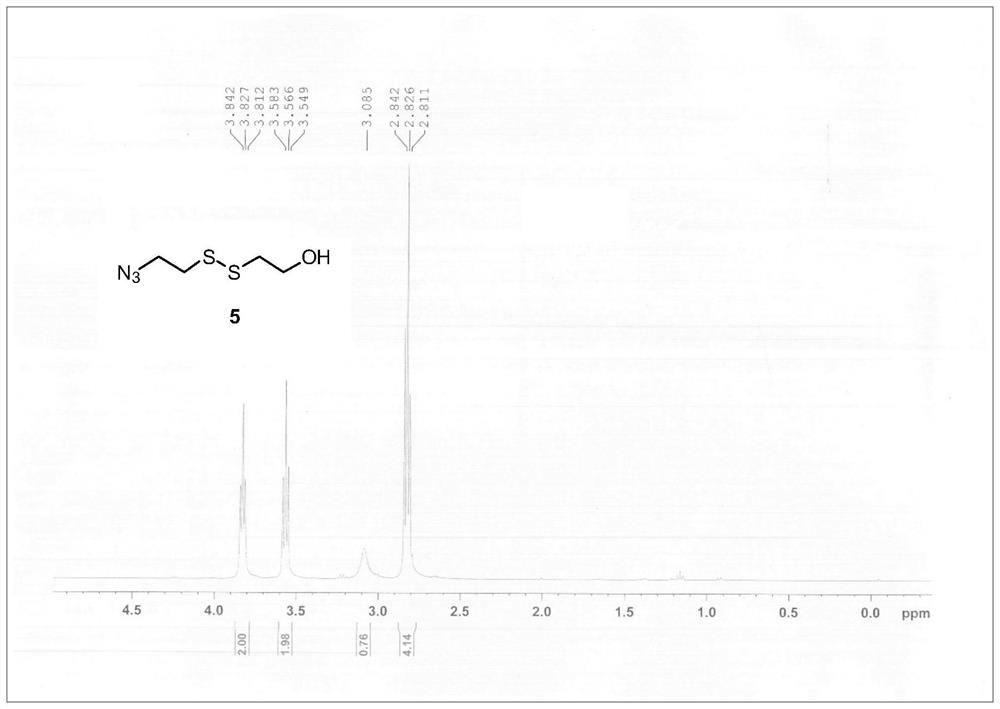
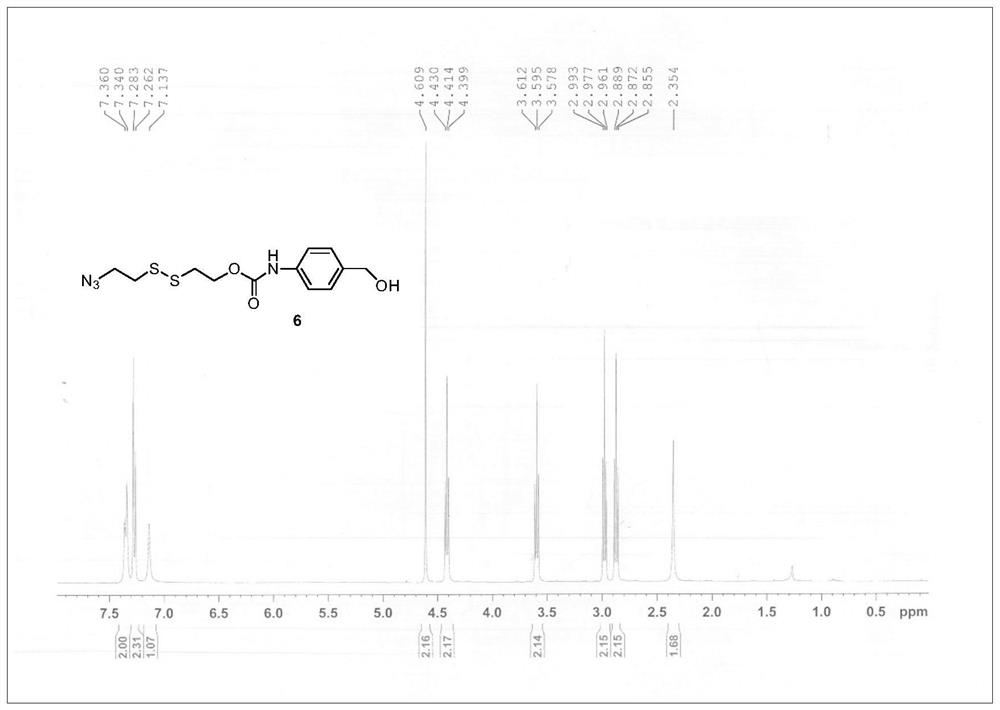
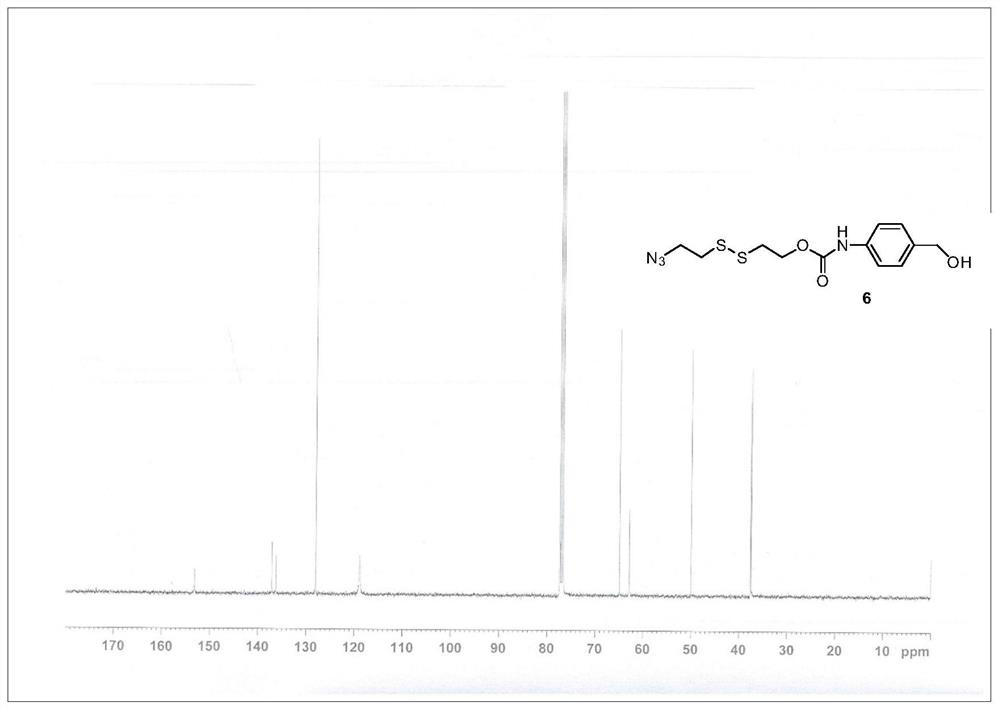
[0061] Step 1. Synthesis of Compound 5. A solution of bis(2-hydroxyethyl)disulfide (2.28 g, 15 mmol) and triethylamine (3.06 mL, 22 mmol) in tetrahydrofuran (THF) (10 mL) was added dropwise 5 mL of methanesulfonyl chloride (1.22 mmol) under an ice bath mL, 15.7 mmol) THF solution, stirred at room temperature for 30 min. The reaction solution was concentrated under reduced pressure to remove THF. The residue was dissolved in 10 mL of N,N-dimethylformamide (DMF), followed by the addition of NaN 3 (2.93 g, 45 mmol), stirred at 60°C. After 6 h, the reaction solution was diluted with 80 mL of water and extracted three times with ethyl acetate (3×25 mL). The organic phase was collected and washed with anhydrous Na 2 SO 4 dry. The solvent was removed by concentration under reduced pressure, and the crude product was purified by silica gel chromatography (petroleum ether:ethyl acetate=6:1) to obtain compound 5 (1.32 g, 4...
Embodiment 2
[0065] Example 2 In vitro drug release studies
[0066] RGD-SS-CA was dissolved in 10% DMF in PBS buffer (50 μM, 500 μL, pH=7.4), and dithiothreitol (DTT) was added to the solution at a final concentration of 250 μM, and incubated at 37°C, respectively. At 30, 60, and 90 min, 50 μL of samples were taken out for HPLC analysis.
[0067] In vitro drug release studies of RGD-VC-CA(3) were performed in the same way. RGD-VC-CA was dissolved in 10% DMF in PBS buffer (50 μM, 500 μL, pH=7.4), cathepsin B was added to the solution, the final concentration was 250 μM, and incubated at 37°C for 4, 12, and 24 h, respectively. A 50 μL sample was withdrawn for HPLC analysis.
[0068] Compared with the cleavage-responsive complex RGD-VC-CA (3), the in vitro release results of RGD-SS-CA showed that ( Figure 8 ), in the presence of the reducing agent DTT, RGD-SS-CA can release 1a quickly and without trace, and at 90 min, RGD-SS-CA can be completely cleaved by DTT, achieving 100% release. W...
Embodiment 3
[0070] Example 3 In vitro study of antiproliferative activity
[0071] 3000 to 4000 human non-small cell lung cancer A549 were seeded in 96-well plates. After 24 hours of inoculation, the medium was removed, new medium was added, and then new medium containing RGD-SS-CA complexes of different concentrations was added to the cells, and blank solvent was used as a control. cells in 5% CO 2 in an incubator at 37°C. After 48h, CCK8 reagent was added and the incubation was continued for 1h. Absorbance at 450 nm was then measured with a full wavelength reader (Thermol Multiskan GO). Each experiment was independently replicated three times. Obtain the value of a in the table below.
[0072] 3000 to 4000 human non-small cell lung cancer A549 were seeded in 96-well plates. 24h after inoculation, remove the medium, add a new medium, first add 1mM GSH-OEt and incubate with the cells, and the cells are incubated in 5% CO. 2 in an incubator at 37°C. Then, after adding RGD-SS-CA con...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More