

Process for the preparation of sodium faropenem
A technology of faropenem sodium and its synthesis method, which is applied in the field of drug synthesis, can solve problems such as heavy metal residues, high price, and limited application, and achieve the effects of improved multi-step synthesis process, simple operation, and shortened reaction time
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

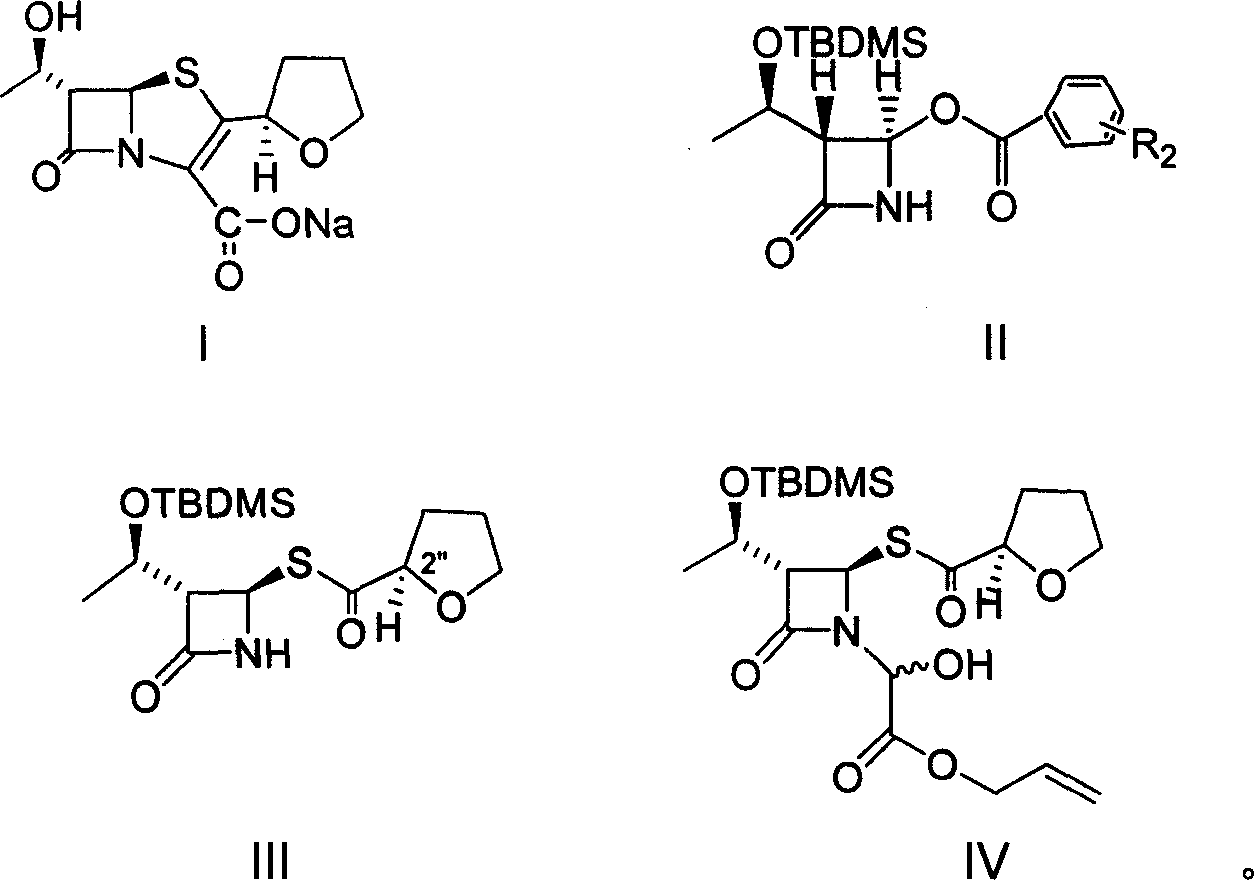
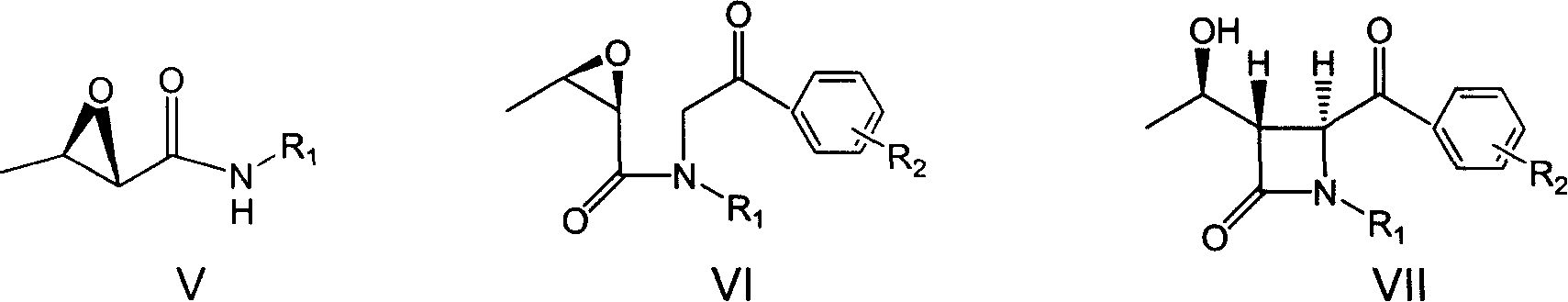
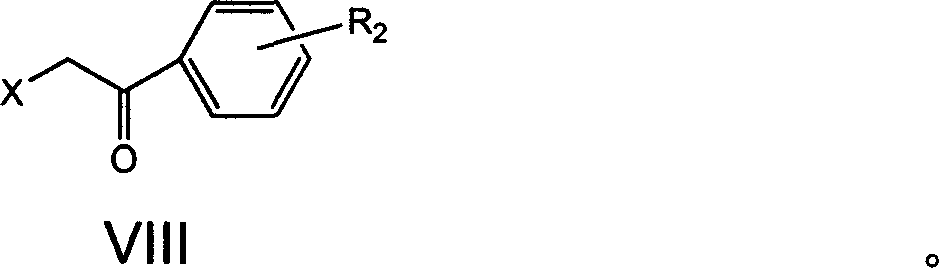
Examples
Embodiment 1
[0039] Example 1: (2S,3R)-2-bromo-3-hydroxybutyric acid
[0040] In a 2000 mL three-neck flask, add L-threonine (100 g, 0.84 mol), potassium bromide (300 g, 2.52 mol), and 2M sulfuric acid solution (1200 mL) in sequence, and cool to 0°C. An aqueous solution of sodium nitrite (90 g, 1.3 mol) dissolved in water (250 mL) was added dropwise, the internal temperature was controlled at 0-5° C., and the dropwise addition was completed. Stir at room temperature for 2 hours. It was extracted with ethyl acetate (300 mL×5), dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to obtain 139 g of the title compound in the form of light yellow viscous. The yield of crude product is 90.4%. The product was directly used in the next reaction without purification.
Embodiment 2
[0041] Example 2: (2S, 3R)-2-bromo-3-hydroxyl-N-(4'-methoxyphenyl)butanamide
[0042] In a 1000mL three-necked flask, (2S, 3R)-2-bromo-3-hydroxybutyric acid (70 grams, 0.38mol), tetrahydrofuran (500mL), and isopropyl chloroformate (47.5 grams, 0.38mol). After cooling to -5°C, triethylamine (38.5 g, 0.38 mol) was added dropwise first, and then a solution of p-methoxyaniline (47 g, 0.38 mol) dissolved in tetrahydrofuran (200 mL) was added dropwise. After the dropwise addition was complete, stirring was continued for 30 minutes. Filter and concentrate. Ethyl acetate (500 mL) was added, followed by washing with 0.5 molar aqueous hydrochloric acid solution (50 mL×3), saturated aqueous sodium chloride solution (100 mL), saturated aqueous sodium bicarbonate solution (100 mL), and saturated aqueous sodium chloride solution (100 mL). Dry over anhydrous sodium sulfate, evaporate the solvent under reduced pressure to obtain 98 g of the title compound as a dark yellow solid, and the yi...
Embodiment 3
[0043] Embodiment 3: (2R, 3R)-N-(4'-methoxyphenyl)-2,3-epoxybutanamide
[0044] In a 1000mL three-necked flask, add (2S, 3R)-2-bromo-3-hydroxyl-N-(4-methoxyphenyl)butanamide (98 grams, 0.34mol) prepared in Example 2, and Sodium (20 g, 0.5 mol), tetrahydrofuran (500 mL). Stir vigorously at room temperature for 3 hours and concentrate. Ethyl acetate (500 mL) was added, washed with water (100 mL) and saturated aqueous sodium chloride solution (100 mL×3) successively. Dry over anhydrous sodium sulfate, evaporate the solvent under reduced pressure, add 100 mL of isopropyl ether, cool, and solidify to obtain 62 g of the title compound as a yellow solid, with a crude yield of 88%. The product was directly used in the next reaction without purification.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More