

Saturation mutagenesis in directed evolution
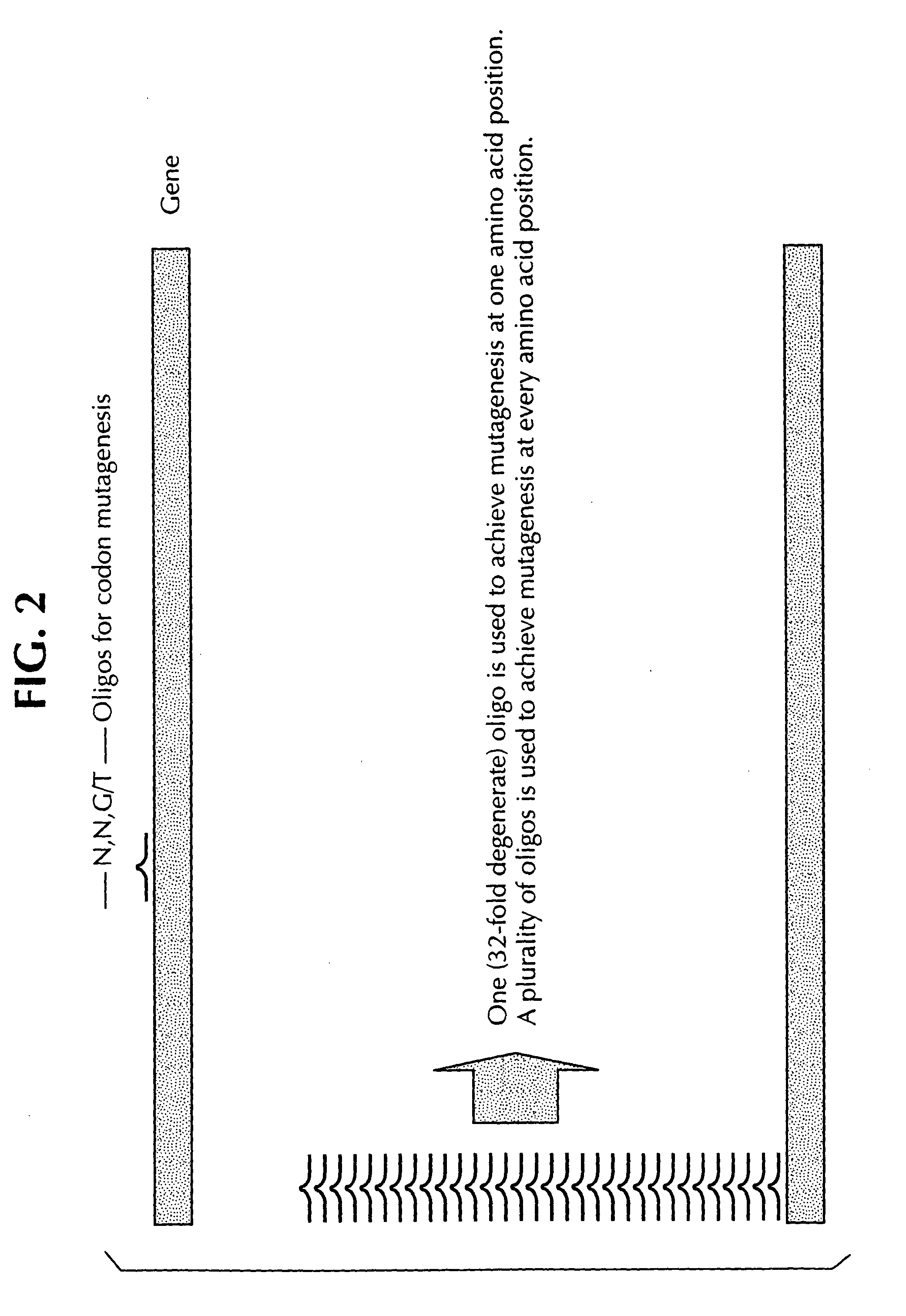
a polypeptide and directed evolution technology, applied in the field of protein engineering, can solve the problems of ineffective site-directed mutagenesis technology, inability to achieve the full range of possible at each position (site) along the polypeptide sequence, and inability to rapidly analyze the large amount of information, so as to reduce the complexity of sequences, enhance biological activities, and enhance the effect of activity
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image


Examples
example 1
Generation of Random Size Polynucleotides Using U.V. Induced Photoproducts
[0335] One microgram samples of template DNA are obtained and treated with U.V. light to cause the formation of dimers, including TT dimers, particularly purine dimers. U.V. exposure is limited so that only a few photoproducts are generated per gene on the template DNA sample. Multiple samples are treated with U.V. light for varying periods of time to obtain template DNA samples with varying numbers of dimers from U.V. exposure.
[0336] A random priming kit which utilizes a non-proofreading polymease (for example, Prime-It II Random Primer Labeling kit by Stratagene Cloning Systems) is utilized to generate different size polynucleotides by priming at random sites on templates which are prepared by U.V. light (as described above) and extending along the templates. The priming protocols such as described in the Prime-It II Random Primer Labeling kit may be utilized to extend the primers. The dimers formed by U.V...
example 2
Isolation of Random Size Polynucleotides
[0337] Polynucleotides of interest which are generated according to Example 1 are are gel isolated on a 1.5% agarose gel. Polynucleotides in the 100-300 bp range are cut out of the gel and 3 volumes of 6 M NaI is added to the gel slice. The mixture is incubated at 50° C. for 10 minutes and 10 μl of glass milk (Bio 101) is added. The mixture is spun for 1 minute and the supernatant is decanted. The pellet is washed with 500 μl of Column Wash (Column Wash is 50% ethanol, 10 mM Tris-HCl pH 7.5, 100 mM NaCl and 2.5 mM EDTA) and spin for 1 minute, after which the supernatant is decanted. The washing, spinning and decanting steps are then repeated. The glass milk pellet is resuspended in 20 μl of H2O and spun for 1 minute. DNA remains in the aqueous phase.
example 3
Shuffling of Isolated Random Size 100-300bp Polynucleotides
[0338] The 100-300 bp polynucleotides obtained in Example 2 are recombined in an annealing mixture (0.2 mM each dNTP, 2.2 mM MgCl2, 50 mM KCl, 10 mM Tris-HCl ph 8.8, 0.1% Triton X-100, 0.3μ; Taq DNA polymerase, 50 μl total volume) without adding primers. A Robocycler by Stratagene was used for the annealing step with the following program: 95° C. for 30 seconds. 25-50 cycles of [95° C. for 30 seconds, 50-60° C. (preferably 58° C.) for 30 seconds, and 72° C. for 30 seconds] and 5 minutes at 72° C. Thus, the 100-300 bp polynucleotides combine to yield double-stranded polynucleotides having a longer sequence. After separating out the reassembled double-stranded polynucleotides and denaturing them to form single stranded polynucleotides, the cycling is optionally again repeated with some samples utilizing the single strands as template and primer DNA and other samples utilizing random primers in addition to the single strands. ...
PUM
| Property | Measurement | Unit |
|---|---|---|
| temperatures | aaaaa | aaaaa |
| temperatures | aaaaa | aaaaa |
| temperature | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More