

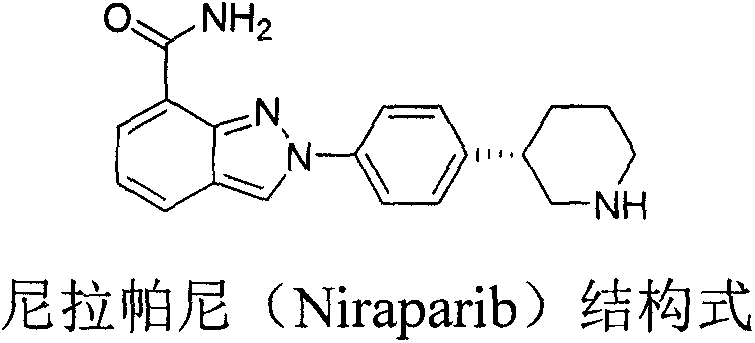
Preparation method of chiral intermediate of niraparib
A chiral intermediate and intermediate technology, applied in the direction of organic chemistry, etc., can solve the problems of complicated enzyme catalysis operation, high production cost, expensive purchase price of enzyme, etc., and achieve the effects of high yield, stable properties and novel route.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0043]Example 1 Synthesis of intermediate I (I-a, I-b, I-c)
[0044]
[0045] The synthesis of I-a: replace good nitrogen in 1000mL eggplant-shaped bottle, add p-bromophenylacetic acid 86g (0.4mol), (S)-4-benzyl-2-oxazolidinone 35.4g (0.2mol), triethylamine 112ml ( 0.8mol) and 350mL toluene, stirred and heated to 80°C, dissolved 49.5ml (0.4mol) of pivaloyl chloride in 300ml toluene, slowly dropped into the reaction solution, after the addition was completed, heated to 110°C, reacted for 14h, TLC showed the reaction completely, cooled to room temperature, filtered, the filter cake was washed twice with ethyl acetate, and the organic phases were combined. The organic phase was washed with water, washed with dilute hydrochloric acid (2M), washed with 5% sodium bicarbonate, dried over anhydrous sodium sulfate, and mixed. Filtration, concentration under reduced pressure, separation by silica gel column chromatography to obtain 62.1 g of white solid (I-a), yield 83%, purity 98%,...
Embodiment 2
[0048] Example 2 Synthesis of Intermediate II (II-a, II-b, II-c)
[0049]
[0050] Synthesis of II-a: In a 2L three-necked flask replaced with nitrogen, add 14.3ml (0.128mol) of titanium tetrachloride and 450mL of dichloromethane, cool to 0 degrees, slowly add 12ml (0.04mol) of tetraisopropyl titanate dropwise , dropwise, after stirring at 0°C for 15 minutes, 31ml of diisopropylethylamine was added dropwise, (0.176). After dripping, stir for 30 minutes. 60 g (0.16 mol) of compound (I-a) was dissolved in 400 ml of dichloromethane and added dropwise to the above reaction solution, and stirred for 2 h after the drop was completed. Then 15.8ml (0.24mol) of acrylonitrile was added dropwise, and the mixture was stirred and reacted at 0°C for 1 day. After the reaction was complete, 1 L of saturated ammonium chloride solution was added, filtered, and the filter cake was washed twice with dichloromethane. The dichloromethane layer was washed with 1M dilute hydrochloric acid (2×50...
Embodiment 3
[0053] The synthesis of embodiment 3 intermediate (III)
[0054]
[0055] In a 2L three-necked flask, 21.3g (0.05mol) of II-a was dissolved in 300mL of tetrahydrofuran, cooled to -10 degrees, and 12ml (0.04mol) of 1M lithium hydroxide solution (100ml) and 1.2ml of hydrogen peroxide were added dropwise in sequence. After stirring for 6 hours at -10°C. After the reaction was complete, methyl tert-butyl ether was added for extraction, the aqueous phase was adjusted to pH 2 with 1M dilute hydrochloric acid, extracted three times with ethyl acetate, the organic layers were combined, and dried over anhydrous sodium sulfate. After filtration, concentration under reduced pressure, and separation by silica gel column chromatography, 12.7 g of (III) was obtained with a yield of 95% and a purity of 97.5%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More