

Preparation method of anti-hepatitis C medicine sofosbuvir
A technology for sofosbuvir and anti-hepatitis C, applied in the field of preparation of anti-hepatitis C drug sofosbuvir, can solve the problems of low total yield of target product, high toxicity of reaction reagents, harsh reaction conditions, etc., and achieves novel routes and equipment. Simple, short synthetic route effects
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

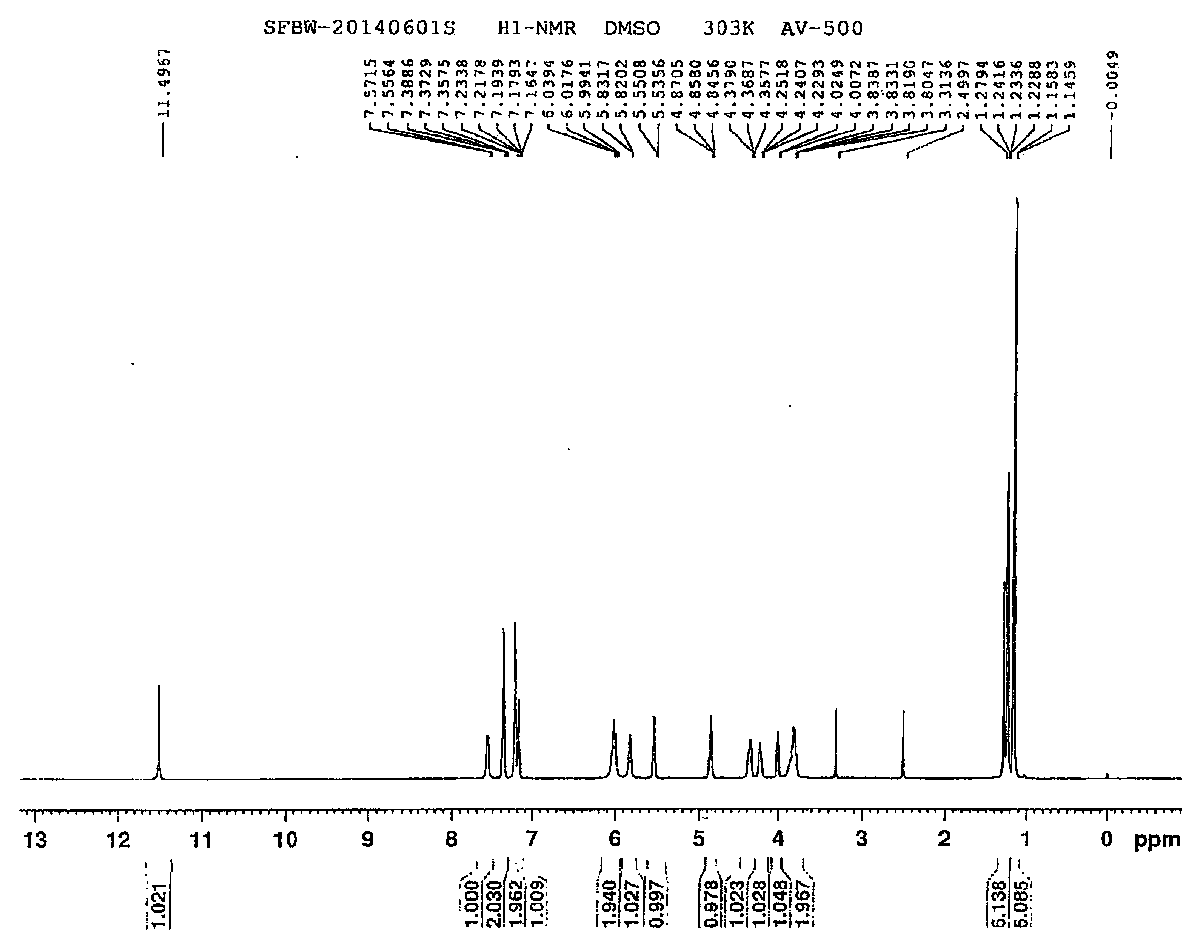
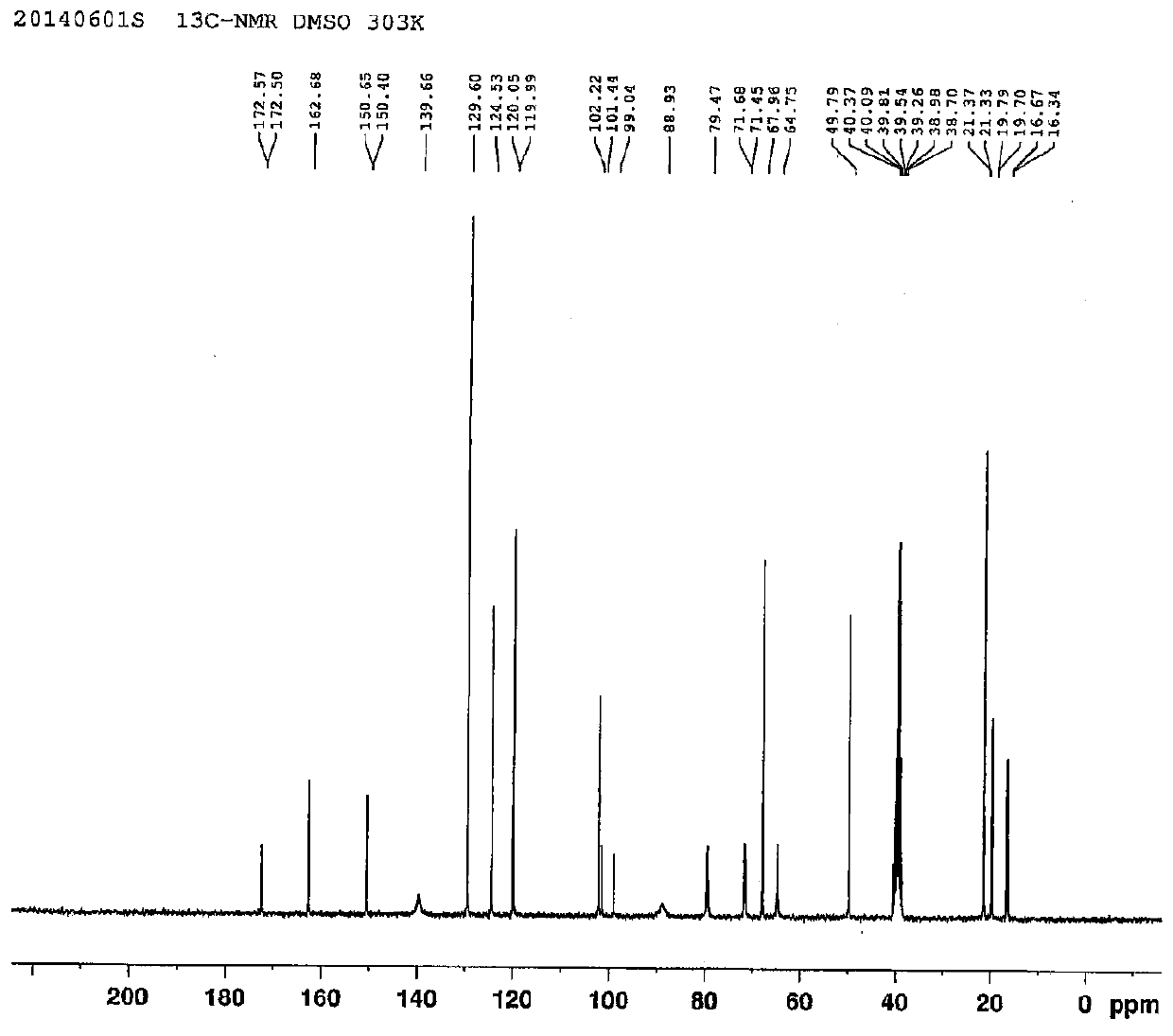
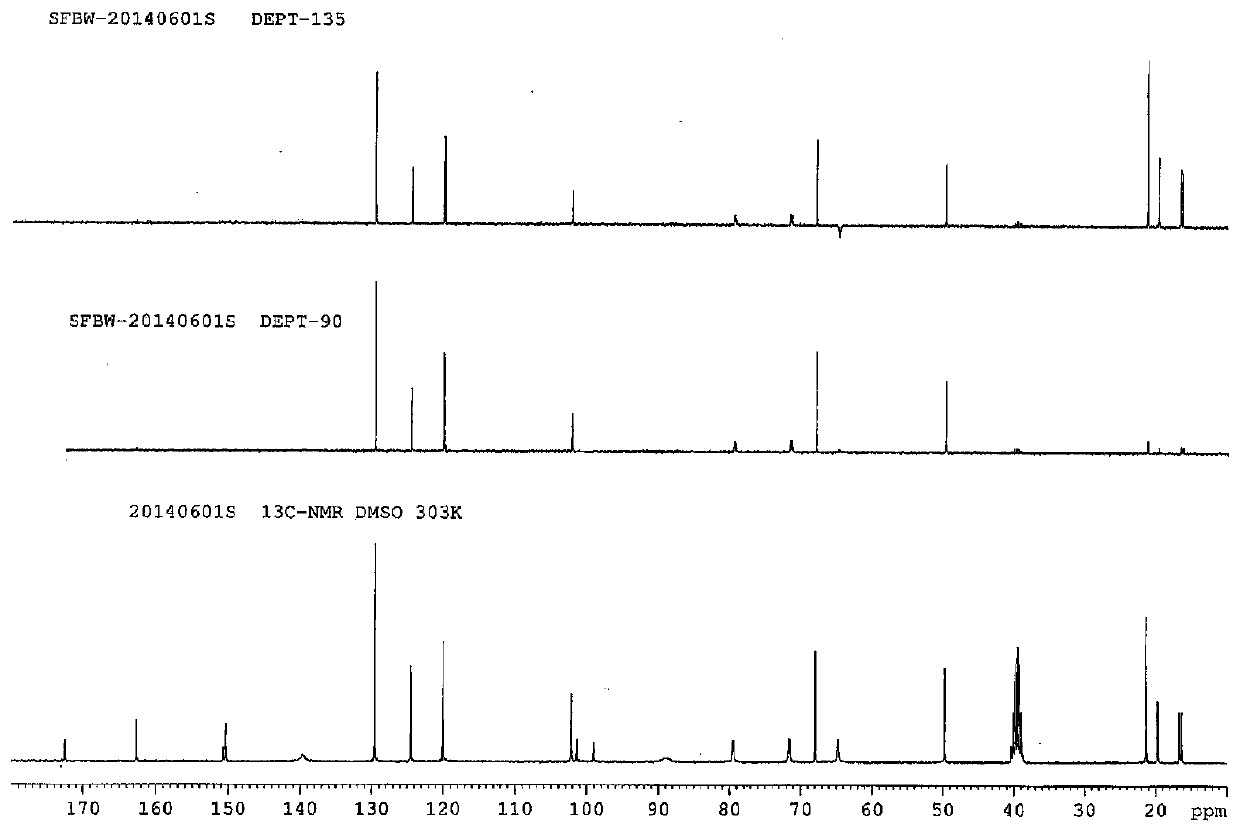
Examples
Embodiment 1
[0053] Example 1: Preparation of 3-((R)-2,2-dimethyl-1,3-dioxolanyl)-3-carbonyl-2-methyl-2-fluoropropionic acid ethyl ester (1 )
[0054] In a 500 ml round bottom flask, add 175 g of β-glycerate ethyl acetone, 200 ml of DMF, 224 g of potassium tert-butoxide, and 130 g of α-fluoroethyl propionate, and the reaction system is heated to 80° for 10 hours. After the reaction, the reaction solution was poured into ice water, extracted twice with 400 milliliters of ethyl acetate, washed with water, dried over anhydrous sodium sulfate, filtered, the filtrate was distilled under reduced pressure to recover the solvent, and the residue was distilled under reduced pressure to obtain 206 g of a light yellow liquid (pressure 5 mm Hg, collection temperature 70-75 °) yield 83%.
Embodiment 2
[0055] Example 2: Preparation of 3-((R)-2,2-dimethyl-1,3-dioxolanyl)-3-hydroxyl-2-methyl-2-fluoropropionic acid ethyl ester (2 )
[0056] Take a 500 ml round bottom flask, add 124 g of intermediate 1, 200 ml of ethanol, cool in an ice bath to 0 °, add 10 g of sodium borohydride in batches, during the addition process, control the temperature not to exceed 5 °, and complete the addition in 30 minutes. After the addition, the system continued to react for 2 hours. After the reaction was completed, 20 ml of dilute hydrochloric acid (1mol / L) was added to the reaction system, and the stirring was continued for 10 minutes. Extracted twice with ethyl ester, washed with water, combined organic phases, dried over anhydrous sodium sulfate, filtered, the filtrate recovered the solvent under reduced pressure, and the residue was distilled under reduced pressure to obtain 113 grams of light yellow liquid (5 mm Hg, collection temperature 90-94 °) , yield 91%.
Embodiment 3
[0057] Example 3: Preparation of 3-((R)-2,2-dimethyl-1,3-dioxolanyl)-3-benzoyl-2-methyl-2-fluoropropionic acid ethyl ester (3)
[0058] Take a 500 milliliter three-necked reaction flask, install a constant pressure dropping funnel, nitrogen protection device, under nitrogen protection, add 62 grams of intermediate 3, 100 milliliters of dichloromethane, 30 grams of triethylamine to the system, and cool in an ice bath to 5 ° below, add 40 grams of Benjia acid chloride dropwise. During the dropwise addition, the temperature does not exceed 10°. After the dropwise addition, the system is warmed up to 40°C for 1 hour. After the reaction, add 100 ml of water to the system for liquid separation. The phase was washed with water, dried over anhydrous sodium sulfate, filtered, and the solvent was recovered from the filtrate under reduced pressure to obtain a yellow solid. Ethyl acetate:petroleum ether=1:1 was recrystallized to obtain 83.1 g of a light yellow solid, with a yield of 94%. ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More