

A kind of preparation method of hepatitis drug intermediate
A technology of intermediates and drugs, applied in the field of preparation of drug intermediates, can solve the problems of expensive, difficult recycling, expensive catalysts, etc., and achieve the effects of less side reactions, high yield, and low requirements for reaction conditions
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

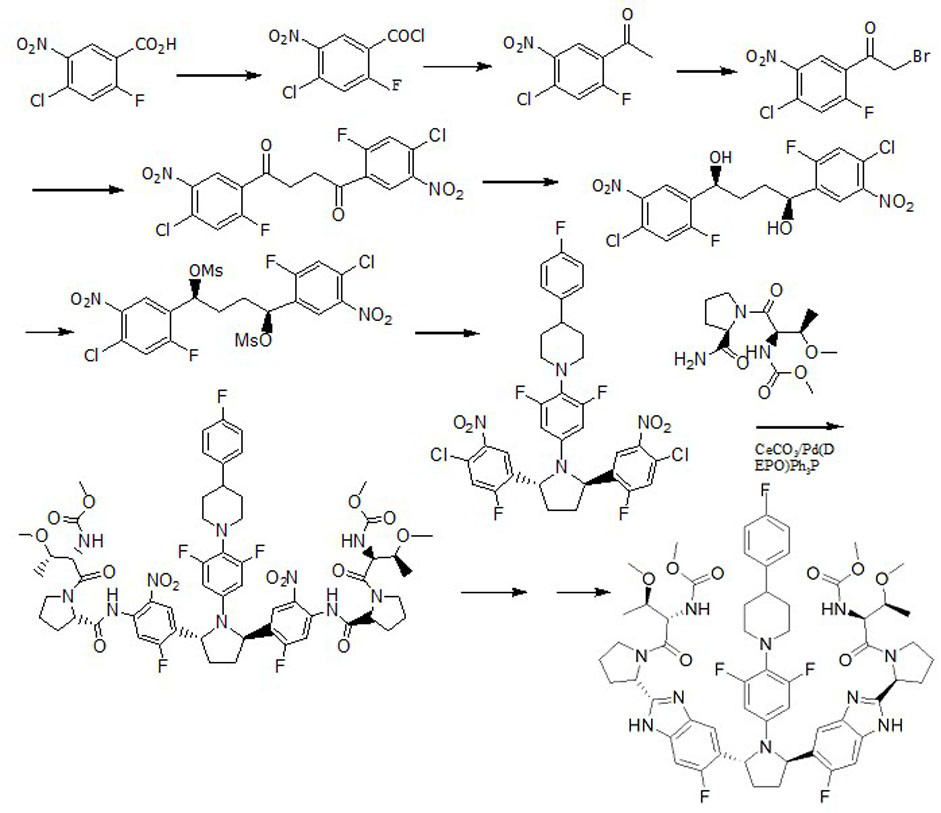
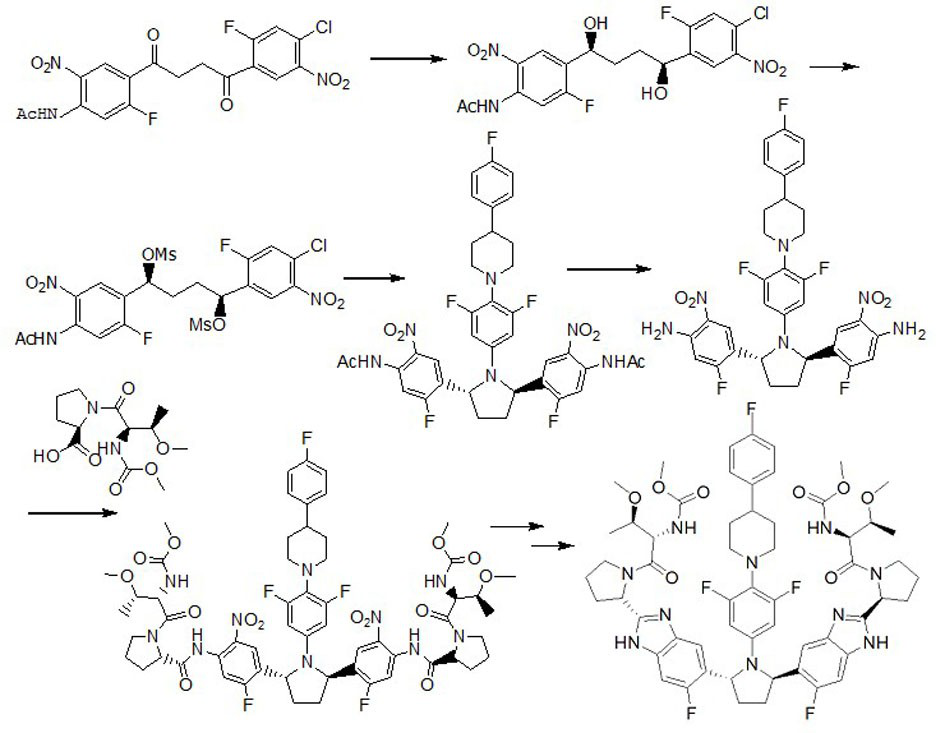
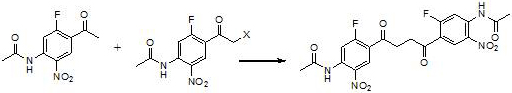
Examples
Embodiment 1
[0039] Embodiment 1: Preparation of 3-fluoroacetanilide
[0040]
[0041] Add 220ml of acetic acid and 55.5g (0.5M) of m-fluoroaniline into a 500ml four-neck bottle, stir to form a homogeneous one, and slowly add 53.6g (0.525M) of acetic anhydride (with obvious temperature rise). After the addition, heat up to 105°C. Insulation reaction 2hrs.
[0042] After the heat preservation is over, remove the acetic acid and acetic anhydride under reduced pressure, and remove to 110°C (-0.095MPa). Slightly cool to 85°C, add 55g of ethyl acetate, stir until uniform, and cool. Cool to room temperature, add 110g of n-hexane dropwise, freeze after dropping, cool to 0°C, stir for 2hrs to crystallize. Suction filtration, washing with n-hexane, and drying to obtain 75.0 g of partial solids, and 3-fluoroacetanilide with a content of more than 99.5% and a yield of about 98%.
Embodiment 2
[0043] Embodiment 2, the preparation of 2-fluoro-4-acetamidoacetophenone
[0044]
[0045] Add 104g of anhydrous aluminum trichloride (0.78M) and 150ml of carbon disulfide to a 500ml four-necked bottle, stir and cool to 0°C, slowly add 39.3g of acetyl chloride (0.5M), after the addition, raise the temperature to 20°C, and stir for 30min . Freeze to 0°C, and add 30.6 g (0.20 M) of m-fluoroacetanilide in batches. After the addition, the temperature was raised to room temperature and stirred for 30 min. Then heated to reflux for 48hrs.
[0046]After reflux, cool to room temperature. Remove the supernatant (to recover carbon disulfide), pour the reaction solution into 500g of crushed ice and 50ml of hydrochloric acid, and stir for hydrolysis. The hydrolyzate was extracted twice with 150ml and 100ml dichloroethane. The extracts were combined and washed twice with 100ml×2 water. The organic phase is a dichloroethane solution of the F-C product.
[0047] The solvent was rem...
Embodiment 3
[0048] Embodiment 3, the preparation of 2-fluoro-4-acetamidoacetophenone
[0049] According to the method in Example 2, nitrobenzene was used instead of carbon disulfide as a solvent. After the reaction was over, it was cooled, poured into crushed ice, stirred and hydrolyzed, and then the organic layer was separated, dried over sodium sulfate, and nitrobenzene was recovered by distillation under reduced pressure. Appropriate amount of methyl chloride, heated to dissolve. Cool to room temperature, slowly add n-hexane dropwise, stir for 1 hr (around 25°C), filter with suction, and wash with dichloromethane / n-hexane (1 / 3). The solid was dried to obtain the Fuke acylation product with a content of more than 97%. Yield 60.1%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More