Methods for preparing 2-alkynyladenosine derivatives
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

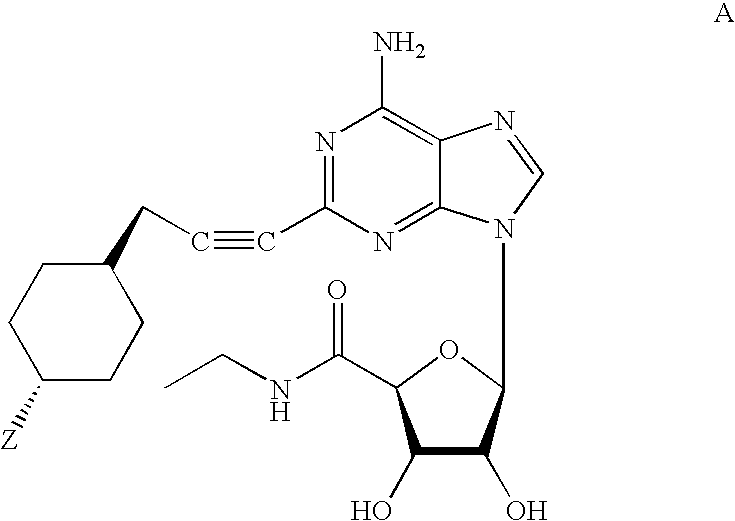
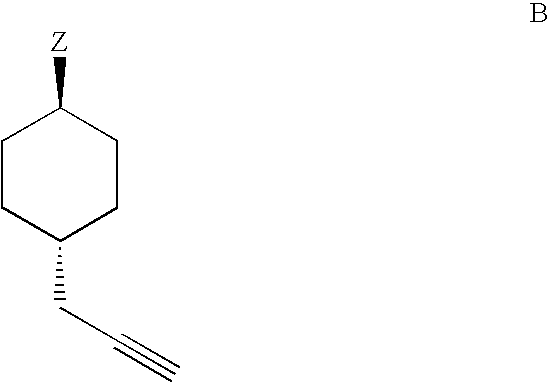
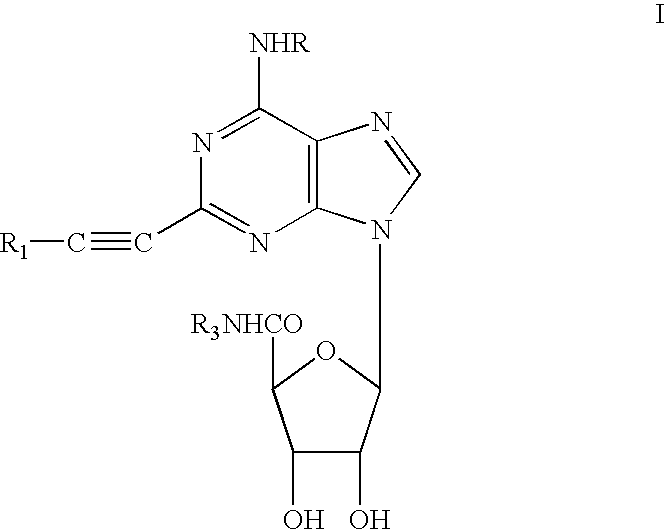
Examples
example 1
Synthesis of [(1R,2R,4R,5R)-4-(6-amino-2-iodopurin-9-yl)-7,7-dimethyl-3,6,8-trioxabicyclo[3.3.0] oct-2-yl]methan-1-ol (Compound 2)
To a suspension of 2-iodoadenosine 1 (10.0 g, 25.4 mmol) in acetone (200 ml) cooled to 0° C. was added dropwise 70% perchloric acid (4.0 mL), resulting in an exotherm of about 5° C. The resultant colorless solution was allowed to warm to room temperature over 30 minutes, then stirred for a further 45 minutes. 1M Na2CO3 (50 mL) was added, resulting in solids precipitating. This was followed by the careful portionwise addition of water (300 mL) with stirring until all solids had dissolved. The mixture was extracted with three portions of CH2Cl2. The combined organics were washed with brine, dried (Na2SO4), filtered, and evaporated to afford Compound 2 (10.26 g, 93%) as a colorless solid. 1H-NMR (600 MHz, DMSO d6): 1.32 (s, 3H), 1.54 (s, 3H), 3.54 (m, 2H), 4.19 (m, 1H), 4.93 (dd, 1H), 5.05 (t, 1H), 5.27 (dd, 1H), 6.05 (d, 1H), 7.74 (bs, 2H), 8.28 (s, 1H);...
example 2
Synthesis of 1′-deoxy-1′-(6-amino-2-iodo-9H-purin-9-yl)-2′,3′-O-isopropylidene-β-D-ribofuranuronic acid (Compound 3)
To a solution of Compound 2 (10.0 g, 23.1 mmol) in CH3CN (200 mL) and water (50 mL) cooled to 0° C. was added iodobenzene diacetate (16.4 g, 50.8 mmol) and TEMPO (0.72 g, 20 mmol). The mixture was stirred at 0° C. for 30 minutes, then allowed to warm to room temperature and stirred for 22 h. The solvents were evaporated and the resulting residue was triturated with n-heptane (400 mL) overnight. The solids were filtered, washed with n-heptane and dried in vacuo to afford Compound 3 (9.80 g, 95%) as an off-white solid. 1H-NMR (600 MHz, CD3OD): 1.45 (s, 3H), 1.65 (s, 3H), 4.78 (d, 1H), 5.50 (d, 1H), 5.67 (dd, 1H), 6.31 (s, 1H), 8.14 (s, 1H); 13C-NMR (150 MHz, CD3OD): 25.45, 27.08, 85.69, 86.09, 88.44, 92.59, 114.92, 120.39, 142.47, 151.08, 157.20, 173.14. LRMS (ES): 448.0 (M+H, 100%).
example 3
Synthesis of N-ethyl-1deoxy-1′-(6-amino-2-iodo-9H-purin-9-yl)-2′,3′-O-isopropylidene-β-D-ribofuranuronamide (Compound 4)
To a solution of Compound 3 (8.0 g, 17.9 mmol) in 50% ethanol / CH2Cl2 (160 mL) was added 2-ethoxy-1-ethocycarbonyl-1,2-dihydroquinoline (EEDQ) (4.65 g, 18.8 mmol) as a single portion. The mixture was stirred at room temperature for 24 hours. Ethanol (80 mL) was added and ethylamine gas (≈45 g) bubbled through the reaction solution over 4 hours. The reaction was stirred at room temperature for 20 hours after which the solvents were evaporated. The resulting solids were dissolved in CH2Cl2 and washed successively with 0.1 M HCl and 1M Na2CO3. The organics were dried (Na2S04), filtered, and evaporated to afford Compound 4 as a pale yellow solid which was recrystallized from CH2Cl2 / hexanes to afford Compound 4 (6.0 g, 71%) as a white solid, m.p. 204-206° C.; 1H-NMR (600 MHz, DMSO-d6): 0.68 (t, 3H), 1.32 (s, 3H), 1.52 (s, 3H), 2.81 (m, 1H), 2.91 (m, 1H), 4.53 (s, 1H),...
PUM
| Property | Measurement | Unit |
|---|---|---|
| Percent by mass | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com